概述
 脊髓小腦性共濟失調
脊髓小腦性共濟失調疾病分類:神經內科
脊髓小腦性共濟失調主要是指Friedreich共濟失調,是脊髓和小腦的變性疾病。Friedreich共濟失調是常染色體隱性遺傳病,主要的臨床表現是共濟失調、弓形足、視神經萎縮、脊柱側彎、心肌病等。
全球患病率約為3/100000。目前尚無特效療法。該群體也被稱為“企鵝人”。
病因
本病的生化改變尚不明。有的病人有丙酮酸氧化的缺陷。脊髓小腦性共濟失調(SCA)是遺傳性共濟失調的主要類型,其共同特徵是中青年發病。血管病、占位病、常染色體顯性遺傳和共濟失調,經CT掃描證實排除其他累及小腦及腦幹的變化。
疾病描述
脊髓小腦性共濟失調是遺傳性共濟失調的主要類型,包括SCA1-21。成年期發病、常染色體顯性遺傳及共濟失調等是本病的共同特徵,並表現在連續數代中發病年齡提前和病情加重(遺傳早現)。
Harding根據是否伴眼肌麻痹、錐體外系症狀及視網膜色素變性分為三型:ADCAI型、Ⅱ型和Ⅲ型,為病人及家系的臨床和基因診斷提供了線索,SCA發病與種族有關,SCA1-2在義大利、英國多見,SCA3常見於中國、德國和葡萄牙。
症狀體徵
1、SCA共同症狀體徵是,30-40歲隱襲起病,緩慢進展,也有兒童期及70歲起病者;下肢共濟失調為首發症狀,表現走路搖晃、突然跌倒和講話含糊不清,以及雙手笨拙、意向性震顫、眼球震顫、痴呆和遠端肌萎縮等;檢查可見肌張力障礙、腱反射亢進、病理征、痙攣步態、音叉振動覺及本體覺喪失。通常起病後10-20年不能行走。
2、除共同臨床表現外,各亞型有各自的特點,如SCA1眼肌麻痹,上視不能較明顯;SCA2上肢腱反射減弱或消失,眼球慢掃視運動較明顯;SCA3肌萎縮、面肌及舌肌纖顫、眼瞼退縮形成凸眼;SCA8常有發音困難;SCA5病情進展非常緩慢,症狀較輕;SCA6早期大腿肌肉痙攣、下視震顫、復視和位置性眩暈;SCA10純小腦征和癲癇發作;SCA7視力減退或喪失,視網膜色素變性,心臟損害也較突出。
疾病病因
常染色體顯性遺傳的脊髓小腦性共濟失調具有遺傳異質性,最具特徵性的基因缺陷是擴增的CAG三核苷酸重複編碼多聚谷氨醯胺通道,該通道在功能不明蛋白和神經末梢上發現的P/Q型鈣通道αDA亞單位上;其他類型突變包括CTG三核苷酸(SCA8)和ATTCT四核苷酸(SCA10)重複序列擴增,在許多病例中這種擴增片斷的大小與疾病嚴重性有關,且年齡愈小,病情愈重。SCAs基因突變改變蛋白的性質,實質無法被正常加工,異常加工的片斷與一種參與非溶酶體降解的缺陷蛋白泛素結合,共同以蛋白酶體的符合體形式轉運至核內,推測這種核內蛋白聚集可影響細胞核的功能。每種SCA亞型基因位於不同的染色體,有不同的大小和基因突變部位,例如,SCA1基因位於染色體6q22-23,基因組跨度450Kb,cDNA長11Kb,含9個外顯子,編碼816個胺基酸殘基組成ataxia-1蛋白,該蛋白位於細胞核,CAG突變位於8號外顯子,,擴增拷貝數為40-83,正常人為6-38。SCA3(MJD)是我國最常見的SA亞型,基因位於14q24..3-32,至少含4個外顯子,編碼960個胺基酸殘基組成ataxia-3蛋白,分布細胞地中,CAG突變位於4號外顯子,擴增拷貝數為61-89,正常人為12-41。儘管SCA有共同的基因突變機制,導致各亞型臨床表現雷同,但仍有差異,如有的伴眼肌麻痹,有的伴視網膜色素變性,病理損害部位和程度也不相同,提示除了多聚谷氨醯毒性作用外,其他因素可能也參與發病。
病理生理
SCA共同的病理改變主要是小腦、腦幹和脊髓變性和萎縮,但各亞型也有其特點,如SCA1主要是小腦、腦幹的神經元丟失,脊髓小腦束和後索受損,很少累及黑質、基底節及脊髓前角細胞;SCA2以下橄欖核、橋腦、小腦損害為重SCA3主要損害橋腦和脊髓小腦束;SCA7的是視網膜神經細胞變性。
診斷檢查
(一)輔助檢查
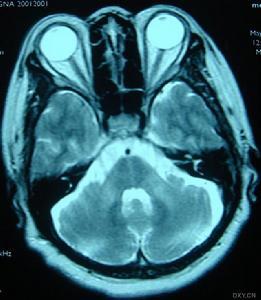
1、CT或MRI顯示小腦萎縮很明顯,有時可見腦幹萎縮;腦幹誘發電位可出現異常,肌電圖顯示周圍神經損害;腦脊液檢查正常。
2、確診SCA及區分亞型可行PCR分析,用外周血白細胞檢測相應基因CAG擴增,證明SCA的基因缺陷。
(二)診斷和鑑別診斷
1、診斷
根據共濟失調、構音障礙、錐體束征等典型共同症狀,以及伴眼肌麻痹、錐體外系症狀及視網膜色素變性等表現,結合MRI檢查發現小腦、腦幹萎縮,排除其他累及小腦和腦幹變性病可臨床確診。然而,臨床僅根據各亞型特徵性症狀、體徵確診仍不準確(SCA7除外),可用PCR法準確判定洲型及CAG擴增次數,進行基因診斷。
2、鑑別診斷
不典型病例需與多發硬化、CJD等引起的共濟失調鑑別。
編輯本段治療方案
本病尚無特異性治療,對症治療可緩解症狀。
(1)左鏇多巴可緩解強直等錐體外系症狀,毒扁豆鹼或胞二磷膽鹼促進乙醯膽鹼合成;氯苯胺丁酸可減輕痙攣,金剛烷胺可改善共濟失調,共濟失調伴肌陣攣首選噓硝安定;ATP、輔酶A、肌苷和維生素B族等神經營養藥可以試用;
(2)手術治療:可行視丘毀損術;
(3)康復訓練、物理治療及輔助行走器械可能有所裨益。進行遺傳諮詢。
治療
對症治療藥物儘管SCA的致病基因各異,但在病變中均累及脊髓和小腦,引起共濟失調等表型,且絕大多數類型致病機制未明,因此目前的治療主要為對症治療,可幫助病人建立自信,改善症狀,在一定程度上延緩病程進展。
脊髓小腦性共濟失調的治療迄今尚無特殊治療,對症治療可緩解症狀,左鏇多巴科緩解強直及其他帕金森症狀,氯苯胺丁酸可減輕痙攣,金剛烷胺改善共濟失調,毒扁豆鹼或胞二磷膽鹼可減輕走路搖晃、眼瞼震顫等,共濟失調伴肌陣攣首選氯硝安定,可試用神經營養藥。手術治療可行事丘毀損術。理療、康復及功能鍛鍊可有裨益。

