
定義
神經內分泌腫瘤
(Neuroendocrine tumor,NET)
神經內分泌腫瘤被定義為高分化(well-differentiated)的神經內分泌新生物,腫瘤細胞形態類似正常腸內分泌細胞,腫瘤細胞排列具有類器官特點,如巢狀、小梁狀或腦回狀等。表達常見的神經內分泌分化標誌物(通常瀰漫和高表達嗜鉻粒蛋白A和突觸素),根據不同發病部位可有相應激素表達(通常是高表達而不需要瀰漫表達)。光鏡下顯示輕至中度的核異形性以及較低的核分裂數(<20/10HPF);根據組織學和增殖指數確定G1和G2。這一定義涵蓋了在2000年WHO分型中的“類癌(Carcinoidtumor)”。
神經內分泌腫瘤是一組起源於肽能神經元和神經內分泌細胞的異質性腫瘤。它不是我們平時所認識的單一的某一種腫瘤,而是一大類腫瘤的總稱。神經內分泌腫瘤可發生於全身許多器官和組織,根據原發腫瘤部位的不同,神經內分泌腫瘤可分為前腸(胸腺、食道、肺、胃、胰腺、十二指腸)、中腸(迴腸、闌尾、盲腸、升結腸)和後腸(遠端大腸和直腸),其中胃腸胰神經內分泌腫瘤最常見。
早期症狀
有功能性的神經內分泌腫瘤常表現為過量分泌腫瘤相關物質引起的相應症狀。
1、類癌綜合徵:突發性或持續性頭面部、軀幹部皮膚潮紅,可因酒精、劇烈活動、精神壓力或進食含3-對羥基苯胺的食物如朱古力、香蕉等誘發;輕度或中度的腹瀉,腹瀉並不一定和皮膚潮紅同時存在,可能與腸蠕動增加有關,可伴有腹痛;類癌相關心臟疾病,如肺動脈狹窄、三尖瓣關閉不全等;其它症狀如皮膚毛細血管擴張症、糙皮病等,偶見皮炎、痴呆和腹瀉三聯征。
2、胃泌素瘤常表現為Zollinger-Ellison綜合徵,腹痛腹瀉常見,呈間歇性腹瀉,常為脂肪痢,也可有反覆發作的消化性潰瘍。
3、胰島素瘤的臨床症狀與腫瘤細胞分泌過量的胰島素相關,特徵性表現是神經性低血糖症,常見於清晨或運動後,其它還有視物模糊,精神異常等表現。
4、胰高血糖素瘤常伴有過量的胰高血糖素分泌,典型表現是壞死性遊走性紅斑伴有貧血以及血小板減少,大約半數患者可有中度糖尿病表現,還可能有痛性紅舌、口唇乾裂、靜脈血栓、腸梗阻及便秘等表現。
5、VIP瘤典型症狀是Verner-Morrison綜合徵,即胰性霍亂綜合徵,表現為水樣腹瀉、低鉀血症、胃酸缺乏症和代謝性酸中毒。
無功能性的神經內分泌腫瘤常缺乏典型的臨床表現,就診時往往已經出現肝轉移。
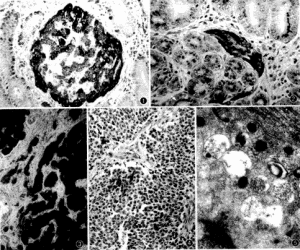 胃神經內分泌腫瘤
胃神經內分泌腫瘤神經內分泌腫瘤較罕見,在全部惡性腫瘤中的比例不足1%,多發生於胃、腸、胰腺,在這類腫瘤中最常見的是類癌,其發生率大約為2.5/100000,占全部胃腸胰神經內分泌腫瘤的50%,根據起源的部位不同,可將類癌分為前腸(肺、支氣管及直到空腸的上部胃腸道)、中腸(迴腸和闌尾)和後腸(直腸),此類腫瘤可發生於整個神經內分泌系統,但最常見的累及部位是胰腺。
臨床表現
1、臨床上短期(6~12個月)出現迅速增長的腫瘤結節分布在原發腫瘤手術區域附近或相應淋巴引流區域的皮膚,且其組織病理形態與原發腫瘤有相似性特別是具有多發性或多灶性瘤灶特徵時,更應考慮為皮膚轉移性癌腫。
2、皮膚或皮下脂肪血管或淋巴管內找到瘤栓癌腫分布構型呈底寬上窄梯形式,一般不與表皮相連,瘤細胞周圍極少有炎性細胞浸潤,無汗腺導管角質護膜分化等,常屬轉移性皮膚腫瘤的特徵。
3、藉助免疫組化標記有助於區別。例如,原發於汗腺來源腫瘤GCDFP-15陽性,而前列腺及甲狀腺轉移到皮膚的腫瘤分別為PSA及TG陽性此外,在臍腹的轉移性皮膚結節必須除外子宮內膜異位或種植性結節還應與卵黃囊或臍尿管胚胎殘留作區別。
4、神經母細胞瘤皮膚轉移癌為散在堅實性,藍色皮下結節無觸痛,可以移動局部刮搓後變白並持續30~60min。類癌腫瘤表現為單個或多發性真皮或皮下結節,可有疼痛。來自Merkel細胞癌的皮膚轉移癌通常為堅實性淡紅色非潰瘍性結節,直徑0.4~0.8cm。甲狀腺癌皮膚轉移癌為肉色或紫紅色皮膚結節常累及頭部和腹部。
主要特徵
 胰腺神經內分泌腫瘤
胰腺神經內分泌腫瘤神經母細胞瘤皮膚轉移癌表現為成簇具有許多核分裂象的小嗜鹼性細胞,形成玫瑰花結狀位於纖細原纖維性嗜酸性基質中類癌皮膚轉移癌位於真皮及皮下脂肪層,由具有圓形核和透明或嗜酸性胞漿,大小和形狀一樣的細胞排列成島嶼狀、巢狀和索狀構成。
Merkel細胞皮膚轉移癌由具有小皰狀核小量胞漿和許多核分裂象的圓形嗜鹼性細胞,成簇排列或形成吻合索或片狀腫瘤細胞大小和形狀一致甲狀腺乳頭狀癌皮膚轉移癌由導管乳頭結構所構成偶有沙瘤體和暗的嗜酸性膠樣物質。
濾泡性甲狀腺癌皮膚轉移癌呈小梁和濾泡狀,伴有腔內膠樣物質。甲狀腺髓癌皮膚轉移癌由片狀多角形或豐滿的梭狀細胞位於纖維血管性基質中構成,基質中常含有淋巴細胞和澱粉樣物質。
Merkel細胞癌具有神經內分泌和上皮分化二者的特點,它們有獨特的核旁和核附近的球狀染色型類似細胞質包涵體和核周圍小圓點,用低分子重角蛋白抗體如AE-1,CAM-5.2和神經細絲染色可以看到。Merkel細胞癌上皮膜抗原,嗜鉻粒蛋白,神經細絲和NSE染色也呈陽性,但S-100蛋白,癌胚抗原和白細胞共同抗原染色為陰性肺轉移性小細胞癌用低分子重細胞角蛋白染色呈瀰漫性核周點樣外觀,用抗神經細絲抗體染色則呈程度不一點狀外觀。
雖然Merkel癌用CEA染色呈陰性反應,但約50%的轉移性小細胞癌CEA染色為陽性反應乳頭狀和濾泡性甲狀腺癌、甲狀腺球蛋白免疫染色陽性,髓性甲狀腺癌、降鈣素染色陽性。
檢查
可通過測定嗜鉻黏多肽A及胰多肽有助於診斷以及腫瘤治療效果監測。診 斷
神經內分泌癌分為類癌(高分化)、不典型類癌(中分化)及小細胞癌(低分化)。
首先根據臨床表現,分為定性診斷(檢測特異性的異常分泌激素水平)和定位診斷(各種影像學檢查)。在各種生化指標檢測中,尿5-羥吲哚乙酸(5-HIAA)對類癌診斷的敏感性為70%,特異性為90%。尿5-HIAA水平升高與腫瘤大小和類癌性心臟病的嚴重程度密切相關,與類癌綜合徵的嚴重程度無關,與腹瀉的發生和預後之間的關係尚不明確。在進行24小時尿5-HIAA測定時,應注意將尿液置於冰櫃或涼爽的地方,以免產生誤差。
鉻粒素A(CgA)水平是神經內分泌腫瘤(NET)的特異性指標,臨床最為常用。在正常人或非神經內分泌腫瘤患者中CgA也有輕度升高,但水平較低。血清CgA的敏感性與腫瘤類型、分化程度及大小有關。且不同實驗室的檢測結果也可能會有所不同。
對於可疑NET但激素水平輕度升高的患者,建議行促胰液素刺激試驗,其敏感性為80%,而鈣刺激試驗的敏感性僅為40%。
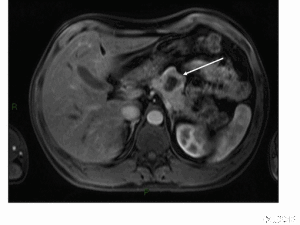
影像學檢查是定位診斷的主要方法,還可同時監測對治療的反應。目前,CT、磁共振成像(MRI)、超聲檢查(US)、內鏡超聲(EUS)等檢查尚未被進一步評價。生長抑素受體顯像的敏感性可根據腫瘤的類型而有所不同,其對於垂體瘤、胃腸胰腺神經性內分泌腫瘤,敏感性超過75%,但對胰島素瘤僅為中度敏感(40%~75%)。
NET確診需要組織活檢及病理學檢測,主要檢測NET的特異性標誌物,同時Ki67(一種增殖細胞相關核抗原)檢測有助於對增殖程度和病變分期的判定。
治 療
可分為生物治療、化療和手術切除。生物治療主要為α干擾素和生長抑素類似物,在分泌相關激素的有功能的NET中應作為首選治療手段,在無功能NET治療中的套用還處於探討階段。目前最新的一種酪氨酸激酶血管生成抑制劑(mTOR抑制劑)已在國外上市,套用於NET的治療。
化療的主要適應證為不能手術的中低增殖度的惡性胰腺內分泌腫瘤,且生物治療失敗。化療方案目前有以下幾種:鏈脲黴素+多柔比星±5氟尿嘧啶、順鉑+依託泊苷、達卡巴嗪,其他還有替莫唑胺、奧沙利鉑、卡培他濱等。鏈脲黴素+多柔比星±5氟尿嘧啶方案最主要的併發症是急性心梗,據報告可達40%。順鉑+依託泊苷主要用於分化較差的NET,但副作用較多,主要見於肝、腎、消化系統和外周神經,每周應監測外周血。達卡巴嗪主要套用於生物治療和鏈脲黴素治療失敗的惡性NET,主要副作用為骨髓抑制。卡培他濱和替莫唑胺聯合套用治療胰腺惡性NET常可收到較好療效,副作用也較小。
作為一種新的治療手段,肽類受體核素放射治療(PRRT)首先要通過國家的許可,治療前要明確腫瘤具有生長抑素受體。其禁忌證包括妊娠和哺乳期婦女、肌酐清除率小於40ml/min、血紅蛋白小於8g/dl,白細胞小於2×109/L、血小板小於75×109/L。由於目前尚無PRRT治療指南,不同地區和臨床機構採用的劑量也有所不同。
影響治療效果的因素有原發腫瘤灶的位置、腫瘤分期和增殖指數,一般來說,原發胰腺腫瘤、Ⅳ期腫瘤和Ki67陽性提示治療效果欠佳。建議每隔3~6個月,對治療反應進行評價,主要檢測鉻粒素A和特異性激素指標的水平,每6個月行一次CT或MRI檢查。
安慰劑對照雙盲隨機前瞻性研究提示,對於腸NET以及有功能或無功能的轉移瘤,長效生長抑素製劑能明顯延長腫瘤患者的中位生存期。在對迴腸和結腸分泌5-羥色胺NET患者2年和5年隨訪中,通過檢測外周血神經激肽A(NKA)和鉻粒素水平可監測腫瘤復發,其中NKA較鉻粒素更為敏感,NKA水平升高提示預後較差,給予干擾素α治療可獲得滿意療效。
可進行手術切除。
預後:經血道或淋巴管轉移的多發性皮膚轉移癌,提示病程已晚期,生存期3~12個月。

