
概述
 進行性骨化性肌炎
進行性骨化性肌炎進行性骨化性肌炎(MOP),又稱進行性骨化性纖維增殖症(FOP)。為一種全身性、進行性、多發性的肌肉骨化現象。並非創傷所致,出生後即有。特點為在胎兒或嬰兒時開始發病,橫紋肌纖維、筋膜、腱膜、韌帶和肌腱由上而下地發生進行性骨化,可同時伴有尺橈骨端、肋骨或脊椎骨附屬檔案的骨端聯合。臨床上可見短頸,少指和小指(趾)的畸形(多見為拇指(趾)的短小)。
國外統計其發病率約為1/200萬,無性別、種族等差異,而據中國國內報導男性略多於女性。截至2013年,全球僅有600名患者診斷罹患此疾。
病因病理
病因
目前(2013年)較公認的該病為結締組織某些成分遺傳方面的缺陷所引起的繼發性鈣化和骨化,屬常染色體顯性遺傳病。該病的遺傳學表現出一定的複雜性和多相性。進來研究比較公認的是其發病與BMP-4關係密切,BMP-4的異常表達被認為是MOP發病的重要因素,BMP-4的過表達是導致活體進行性異位骨化這一連鎖反應的獨立觸發因子。
其致病候選基因已成功定位於2q23-24的ACVR1基因,該基因全長約138.6kb,含有11個外顯子(外顯子1和2僅含5’UTR,蛋白起始位點在外顯子3)。
至21世紀初,全世界僅發現600-700例,對其中約40例進行了基因診斷,均發現為ACVR1基因的4號外顯子存在G617A(R206H)點突變,未發現遺傳異質性。
病理改變
患者肌肉本身無異常,主要累及肌肉的結締組織。早期有明顯間質內水腫和結締組織內增殖,肌纖維發生繼發性萎縮和變性,後期有中胚層組織鈣化和骨化。
臨床表現
病變在嬰兒開始,多在6歲前起病,偶有在出生之後即見肌腱異常。平均發病年齡為4.92歲,男女比例報導不一。其臨床表現有兩個特點:
 進行性骨化性肌炎
進行性骨化性肌炎1、先天性拇指畸形
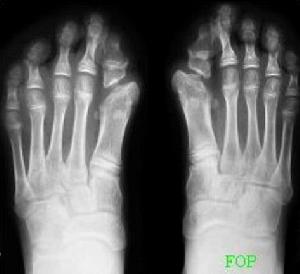
為該病最早的特徵表現,幾乎所有的患者都會發生。手足畸形為指(趾)骨短小。拇趾較拇指為多見,有的表現為一節指骨(趾骨)的缺如,或二趾間融合變短,也可見掌骨和近節趾骨骨性聯合。
2、進行性軟組織異位骨化。
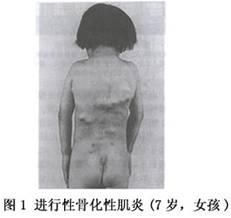
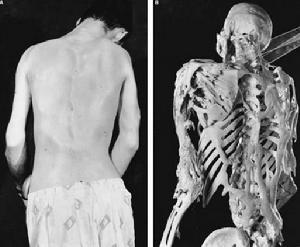
異位骨化多發生於肌腱、韌帶、筋膜和骨骼肌等處,其蔓延特點為從頭向尾、從背側向腹側、從中線向四肢發展,骨化可由外傷、感染和手術等誘發加速。
早期表現為局部包塊紅腫熱痛,繼而包塊逐漸骨化,局部組織攣縮,關節僵硬,逐漸強直僵硬以致活動障礙,如頸的前屈和顳頜關節活動受限,最終肢體運動功能喪失。
一般30歲左右髖關節融合致行走功能喪失,下頜功能障礙致進食不能而全身衰竭。患者大多死於衰竭或感染。
診斷檢查
凡每一個病例均有對稱性拇指(或拇趾)畸形,對診斷本病很有幫助。
生化檢查
一般常規實驗室檢查無特異發現,血生化檢查可發現磷酸酶較高。
心電圖
由於心肌纖維的發育不良,在心電圖上可有異常改變,胸部肌肉受到廣泛的侵犯則引起呼吸衰竭。
X 線片
X 線片顯示腫物的軟組織當中有分散鈣化影。過一段時間,急性期的症狀和體徵消失後,腫物變小,鈣化影也縮小,但密度增高。X線片上可見柱狀或不規則形態的團塊狀不同密度的骨化陰影,可與骨骼相連,也可完全游離。骨骼呈現失用性萎縮。
治療方法
 進行性骨化性肌炎
進行性骨化性肌炎截至21世紀初,尚無特殊的有效療法,應避免受傷受寒,或可防止惡化。可試用腎上腺皮質激素,但療效未定。
病變比較局限者可考慮切除,但有時手術的創傷可加劇病變,導致更多的骨形成。
病變活動期的治療原則是受累部位完全制動休息,不必進行任何物理治療。急性期過後再逐漸恢復活動。
肘關節附近的骨化性肌炎多為積極活動所誘發。預防的辦法是傷後除輕柔自主練習外,不做任何理療。肌炎的病變成熟後,如功能不受限制則不必處理。若殘留妨礙活動的小病變,超過急性期1年,X線照片上肯定為成熟骨組織者,方可手術切除。有的作者主張在急性期做放射治療,醫學界多持相反意見。
預後及預防
發生併發症者影響肢體功能,病變局限者,預後尚好。
因病因尚不明確,目前無系統預防措施,應參照遺傳病學預防方法,做好遺傳病學諮詢工作等 。
病例
 納莫克
納莫克英國西倫敦的女孩納莫克,2013年17歲,罹患了罕見的“進行性骨化性肌炎”,目前(2013年7月),她的手臂關節目前已被“鎖”在腰部位置,而彎曲的手臂再也無法自由移動,就像靜止的雕像一般。納莫克的肩膀也長出了多餘骨骼,她的手臂再也無法舉至腰部以上,在沒有人協助的情況下,她連自行刷牙、洗頭和更衣都有困難,而病情卻只會每況愈下。即使是輕微的碰撞都會令她感到劇烈疼痛,而疼痛會引發骨骼增生,使她的身體關節逐一死鎖。
納莫克無法接受注射或活組織切片等檢查,因為這些治療只會使她體內增生更多多餘骨骼。由於進行性骨化性肌炎還沒有任何治癒的方法,納莫克必須仰賴無處方的止痛藥來對抗病魔。

