
病因
 家族性澱粉樣多發性神經病變
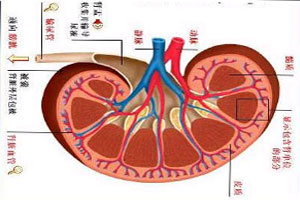
家族性澱粉樣多發性神經病變這些沉積的纖維性蛋白,會集結在臟器、血管、心臟、腸胃系統,特別是周邊神經系統(如神經乾、神經叢、感覺運動神經、自律神經等)而導致不同的症狀與病變,發病年齡分布從20歲到70歲,平均發病年齡為30歲左右。
發生率
關於發生率,截至21世紀初尚未有確切的統計,依不同的疾病類型與種族,疾病的盛行率也有所不同。在葡萄牙北部TTR類澱粉沉積症(TTR amyloidosis)的發生率為1/538,但是在歐洲裔的美國人中,其發生率則為1/100,000;心臟型的TTR 類澱粉沉積症在非洲人中很常見,據統計非裔美國人的發生率約為3-3.9%,在西非的某些區域其發生率為5%。臨床表現
 家族性澱粉樣多發性神經病變
家族性澱粉樣多發性神經病變神經型的TTR類澱粉沉積症會侵犯周邊神經和自律神經系統,造成周邊神經病變和身體控制的困難。相關的症狀包含如腹瀉、陽痿、便秘、排尿方面的問題和姿勢性低血壓,眼睛也容易發生如玻璃體溷濁、乾眼症、青光眼等病變。有些患者還會有心臟和腎臟的問題,或發生腕隧道症候群(手和手指產生刺麻和無力感)。神經型的患者又可依好發的種族分為第一型FAP和第二型FAP。
第一型FAP的患者(FAP-I好發於葡萄牙、瑞典、日本),其神經病變的進程是緩慢的,通常是從下肢末端的感覺神經開始出現異常如皮膚感覺異常,溫覺和痛覺異常較早出現,震動和姿勢感覺異常較後出現,幾年之後運動神經也會跟著產生病變。通常在30~40歲後此疾病會開始發展,但是真正發病的年齡可能會更晚。
第二型FAP的患者(FAP-II好發於印第安那-瑞士; 馬里蘭州-德國),開始的症狀常以腕隧道症候群來表現,感覺運動神經和自律神經的病變與內臟方面的症狀一起出現。
軟腦膜型TTR類澱粉沉積症一開始會侵犯中樞神經系統,且類澱粉常會沉積在軟膜蜘蛛膜中(為兩層薄薄的組織可包覆腦部和嵴髓),逐漸增加沉積物可能會造成中風或腦部出血、水腦、運動性共濟失調、肌肉僵硬或無力、痙攣、失智症等。其他類似神經型TTR會出現的眼睛症狀亦可能發生,這一類患者又可稱為眼睛軟腦膜型(oculoleptomeningeal form)。這類患者的腦嵴髓液中常可發現高量的蛋白沉澱,釓顯影劑促進磁振造影(gadolinium-enhanced MRI)可以顯示出腦部、腦室和嵴髓的表面有廣泛性的增厚。
心臟型最易被侵犯的位置為心臟,常見的症狀如:心律不整、心臟肥大等,這些異常會導致漸進式的心臟衰竭或致死。有時候心臟型的患者亦可能發生輕微的周邊神經病變。這一型的患者通常較晚發病,大約在50歲後才會陸續出現症狀,且通常以窄縮性心肌症出現。
遺傳模式
此疾病為體染色體顯性遺傳,最常見的致病基因TTR位於第18號染色體上的18q11.2-q12.1位置,已在TTR基因上找到將近100個相關的突變點位與缺損(deletion),其中以Val30Met為最常見的突變(第30個胺基酸由原本的纈氨酸突變為甲硫胺酸),且分布於不同種族間。
TTR類澱粉沉積症的患者約有1/3遺傳自父母,另外2/3則為自發性的突變。若為異型合子的患者,下一代罹患此症的機率為50%;若為同型合子的患者,其手足帶有1個突變基因的機率為50%,帶有2個突變基因的機率為25%,下一代則都會帶有突變的基因。
診斷原則
在診斷上,可以藉組織切片以剛果紅(Congo red)染色,觀察有無類澱粉的沉積,若為陰性反應,可以藉免疫組織化學法(immunocytochemical study)來進一步確認。此外,還可以利用質譜技術檢測血清中是否有TTR蛋白的相關產物。
針對TTR類澱粉沉積症的患者,可利用基因檢查來協助確診,TTR基因有4個外顯子(exon),致病相關的基因分布在外顯子(exon)2,3和4上,基因序列分析可以檢測出近99%的致病基因。
治療方法
 家族性澱粉樣多發性神經病變
家族性澱粉樣多發性神經病變由於TTR大部分是經由肝臟而生成,所以對於神經方面的病變,肝臟移植是目前(21世紀初)唯一較有效的治療方式。患者如有以下條件,通常會被建議進行肝臟移植治療:
1.小於50歲;
2.受疾病侵犯的時間小於5年;
3.多發性的神經病變只局限於下肢或自律神經系統;
4.沒有嚴重的心臟或腎臟病變。另外,患者需要定期以神經傳導檢測,追蹤其運動神經病變的情形。其它家族中的成員,若已提早經基因檢查確定診斷,亦可以提早確定治療方針。
疾病預後
對於TTR類澱粉沉積症患者的預後,會依據不同的突變而有所不同,影響患者生存能力較主要的症狀有:
大腸機能障礙會產生嚴重的腹瀉導致體重流失;因鬱血性心臟衰竭而使肺部水腫,最後導致呼吸困難;少部分患者會發展為腎臟衰竭,需要以腎臟透析治療。從患者出現症狀開始算起,其存活期間可能為5年、10年或15年,甚至有患者可以再存20年。

