
概述
【疾病類別】斑色魚鱗癬(Harlequin ichthyosis)
【疾病名稱】斑色魚鱗癬(Harlequin ichthyosis)
【現階段政府公告之罕見疾病】是
【是否已發行該疾病之宣導單張】否
病理簡介
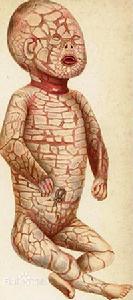 斑色魚鱗癬
斑色魚鱗癬HI的致病主因為位於2q34的ABCA12基因突變,使調控脂質運送的ABCA12蛋白異常所致。皮膚表皮又可細分為5層,角質層為最外層,由死掉的角質細胞堆積而成。活的角質細胞中有富含脂質的層板顆粒(lamellar granules),ABCA12為層板顆粒膜上的蛋白質,負責將脂質運送至顆粒中。顆粒中的脂質最後會經胞釋作用(exocytosis)充滿細胞間隙。因此在角質層中還有脂質層(lipid lamellae)填充在死掉的角質細胞間,和皮膚的脫屑與防水作用有關。
HI患者的角質細胞,其層板顆粒缺乏或有異常的ABCA12蛋白,使角質層喪失脂質,皮膚脫屑作用無法進行,所以造成角質大量堆積、皮膚滲透性異常。這些大規模、角狀、緊實的角質斑塊,會牽扯深沈組織,在皮膚上產生龜裂性的傷痕,這些如盔甲般的角質塊也使眼部、嘴部、耳朵與四肢不正常緊縮,破壞皮膚的保護與防禦性,使水分、體溫的調控失衡,常並發嚴重的細菌感染。
遺傳模式
常染色體隱性遺傳。HI為罕見的嬰兒疾病且致死率高,並發性感染為主要死亡原因。但近期因良好的照顧,嬰兒期的存活率已提升,男女或種族的患病率目前並無特殊統計。
臨床症狀
此疾病主要病變在皮膚,但其他系統也會受到影響,常伴隨肢體變形的情形,患病的嬰兒大都有早產的現象。眼瞼外翻和嘴唇外翻為典型表征,其他症狀為耳廓發展不良或沒有,手臂、足部、手指、腳趾彎曲或發育不全,而皮膚障蔽的破壞會導致脫水、電解質失衡、體溫失調、感染機率增加。堅硬、像盔甲般的角質疤塊會限制呼吸,使進食受限,但患者小腸的吸收功能正常。
各器官之症狀一覽表
皮膚:一出生就可見患者全身的皮膚遍布大且厚的角質化斑塊,並以深而紅的裂痕為斑塊的分界。
眼睛:上下眼瞼嚴重外翻,使眼睛乾燥與增加外傷的可能性。
耳朵:耳廓因發展不良,耳朵較正常人小或甚至沒有。
嘴唇:因皮膚嚴重角質化而牽引嘴唇嚴重外翻,使嘴唇無法閉合。
鼻子:發展不全,常有鼻翼消失症狀。
運動:四肢被高度角質化,被厚厚的皮膚所包圍,導致手臂、腿、手指、腳趾活動受限,有四肢蜷曲的現象;手指、腳趾、指甲發育不良。
體溫:角質化皮膚使汗腺功能受到影響,有失溫的情形。新生兒對溫度的適應能力差,常有體溫過高溫的情形。
呼吸:呼吸受限,嚴重者伴隨呼吸衰竭。
水份調控:水份流失嚴重,因脫水造成心跳過速或少尿。
中樞神經系統:過厚的角質層造成由中樞神經系統調控的自主性運動受到影響,或因全身代謝異常造成癲癇,有時中樞神經的症狀,也可能導因於感染或組織缺氧。
相關診斷
超音波產檢、臨床表征、基因診斷、皮膚組織切片。
1.臨床診斷:臨床表征明顯,為診斷依據。
2.皮膚切片:以皮膚免疫組織染色法,可觀察角質細胞中的層板顆粒有無異常的現象。
3.基因檢查:2q34的ABCA12是否有突變。
4.產前診斷:若家族中缺陷基因已經基因檢查確認,可於懷孕10-12周時以絨毛膜或懷孕14-16周時以羊水直接進行胎兒的基因分析;另外,產撿或可藉由超音波檢查觀察到胎兒皮膚的異常。
治療預後
患者的壽命未有正確統計,曾有活至19歲的HI患者被報導過,突發性敗血症為新生兒患者最常見的死因,目前只能以症狀治療。存活的患者智力大都正常,但發展落後,且身材短小,需定期追蹤其甲狀腺功能和生長發育狀況,部分患者會伴隨類風濕性關節炎。
1、體內恆定:新生兒的呼吸、循環恆定是很重要的,因此需在保溫箱中,隨時監測體溫、呼吸、心跳及血氧濃度,避免體溫過高。靜脈注射通常都是必要的,以維持身體水份與電解質的平衡。
2、眼瞼外翻:可擦凡士林以保護眼睛。
3、角質皮膚:可幫患者一天泡兩次澡,或用沾生理食鹽水的紗布輔以無刺激性的軟膏,幫助角質軟化和脫皮作用,但不建議使用水楊酸(Salicylic Acid) 等去角質劑(Keratolytics)。
4、避免感染:儘量讓患者在在無菌環境,以避免細菌感染,並密集做皮膚細菌培養,監測是否有金黃色葡萄球菌、綠膿桿菌等的感染。

