
簡介
 先天小瞼裂綜合症
先天小瞼裂綜合症 分型
先天性小瞼裂綜合症有家族遺傳性的特徵。
Ⅰ型:普通型:由父親傳代,女性患有不孕症,外顯完全,為100%。
Ⅱ型:父母親傳代機會相等,不完全外顯,外顯率約為96.5%
病因
在遺傳的變異上,約有半數(50%)的患者為無家族史的偶發案例,主要為第三對染色體長臂23(3q23)位置上的FOXL2(forkheadtranscriptionfactor2)基因突變所導致。此外,相關研究發現,3q23染色體位置上的微小片段缺失(interstitialdeletion/ microdeletion/ submicroscopicdeletion),及3q2位置上平衡型轉位(balancedtranslocationinvolvingband3q2)的染色體變異,也都與此疾病的發生有關。
症狀
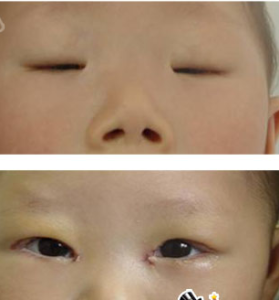
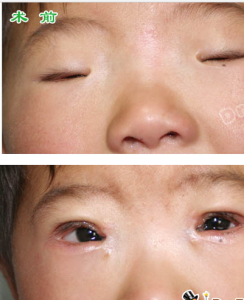 先天小瞼裂綜合症術前,術後對比
先天小瞼裂綜合症術前,術後對比 第一型女性患者的初經來潮通常正常,但因早發性卵巢衰竭,而導致續發性的經量過少或無月經,並將造成不孕的問題;而患者的其他第二性徵發育通常正常。第二型患者則沒有上述表征。
無論第一型或第二型患者,在出生時通常即表現出複雜的先天眼瞼畸形;由於眼瞼發育不良(缺了眼皮上自然摺疊的薄皮膚)、邊緣呈S-型的上眼瞼及眼瞼外翻,使得眼瞼的裂縫(瞼口)狹小,及內眥贅皮反轉,而可能因為上眼瞼提肌的缺失或萎縮,而導致眼皮下垂。
此外多數患者具有眼距過寬(telecanthus)、寬鼻樑(broadnasalbridge)、低位耳(low-setears)及短小的人中(shortphiltrum)外觀特徵。偶會出現眼球震顫(Nystagmus)及小眼症(microphthalmos)的臨床表征。而鼻淚管的發育不良,有時會造成患者排出淚水的淚點(lacrimalpuncta)狹窄。
Dawsonetal(2003)針對204名BPES個案的視力問題予以統計,發現其中有斜視(strabismus)問題者占了20%,折射異常(significantrefractiveerror)占了34%,而雙側及單側弱視則各有21%及20%。
多數患者可能出現智慧型及發展上的遲緩,而因基因突變而致病的患者,通常有正常的智力發展。
診斷
在診斷上主要依患者外觀上的臨床表征,並配合相關的眼科檢查;包括眼瞼縫隙(寬度及高度)的測量、視力及眼球折射檢查,以及斜視與眼球活動度等評估,以進行診斷。
而第一型女性患者需接受生殖系統的理學檢查(如:骨盆腔超音波,以評估子宮及卵巢的發育情形),及相關內分泌數值(如:雌性素、黃體素等)的檢測,以評估其生殖功能。
部分患者可經分子生物技術,以基因檢測及染色體微小片段缺失的檢查,找到突變基因或與疾病相關的染色體變異;
在基因檢測上,約有70%的患者可經基因序列分析(Sequenceanalysis),檢測出FOXL2的基因突變。
在染色體微小片段缺失的檢測上,約有10%的的患者可以多重連線探針擴增法(MultiplexLigation-dependentProbeAmplification; MLPA),找到微小片段的缺失。
此外,5%的患者可借衛星區域及核甘酸多形性分析(MicrosatelliteandSNPanalysis),發現特定的基因外重組(Extragenicrearrangements)所致的突變;但這部分的檢測仍為研究階段,僅作為臨床診斷上的參考。
治療
醫療團隊的照護包括:遺傳專科、眼科、整形外科等各專業的協同合作,透過各項評估檢查及治療計畫,將能使患者因疾病所致的症狀,獲得最佳的改善。
在瞼口狹小、內眥贅皮反轉,及眼皮下垂等外觀及視力問題的處理上,通常建議患者需即早接受眼科的評估,若有礙視力或基於外觀上的考量,可考慮於適當年齡,接受相關的外科整型及成形手術。
出現早發性卵巢衰竭的第一型患者,可接受女性激素的荷爾蒙治療,而患者的治療劑量,將依其年齡、荷爾蒙數值及相關生理評估而定。
患者若出現發展及智慧型上的遲緩,可接受發展上的評估,以即早接受肢體或智慧型發展上的早期療育,使患者能獲致與一般同年齡兒童的發展水平,而能順利就學接受教育。

