
疾病概述
 發病機理
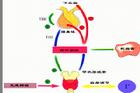
發病機理先天性腎上腺皮質增生症(Congenitaladrenalcorticalhyperplasia,CAH)是由於腎上腺皮質激素生物合成酶系中某種或數種酶的先天性缺陷,使皮質醇等激素水平改變所致的一組疾病。屬常染色體隱性遺傳病,引起男性化者又稱腎上腺性徵異常綜合徵。
每15000嬰兒中可能就有一個患有這種疾病,幾乎所有的新生兒都要接受檢查。倘若沒有檢測到這種疾病存在,它會嚴重影響嬰兒在出生後數周的身體健康。這種酶缺陷還會導致皮質醇激素的缺失,後者會影響心臟問題,以及腎上腺產生的雄激素的上升。
病因
腎上腺合成皮質醇是在垂體分泌的ACTH(促腎上腺皮質激素)控制下進行的,先天性腎上腺皮質增生症時,由於皮質醇水平降低,負反饋作用消除,抑制垂體釋放ACTH的作用減弱,致ACTH分泌過多。其臨床表現和生化改變取決於缺陷酶的種類和程度,可表現為糖、鹽皮質激素和性激素水平改變和相應的症狀、體徵和生化改變,如胎兒生殖器發育異常、鈉平衡失調、血壓改變和生長遲緩等。
典型的CHA發病率約為10/10萬,而非典型的發病率約為典型的10倍,並有種族特異性。
症狀體徵
21—羥化酶缺乏症(ZD—OHD)
 先天性腎上腺皮質增生症機理
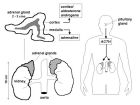
先天性腎上腺皮質增生症機理 (1)單純男性化型(SV)
系21—羥化酶不完全缺乏所致,酶缺乏呈中等程度,11—脫氧皮質醇和皮質醇、11—脫氧皮質酮等不能正常合成,其前體物質17—羥孕酮、孕酮、脫氫異雄酮增多,但仍可合成少量皮質醇和醛固酮,故臨床無失鹽症狀主要表現為雄激素增高的症狀和體徵。
女孩表現為假兩畸形:由於類固醇激素合成缺陷在胎兒期即存在,因此,女孩在出生時即呈現程度不同的男性化體徵,如陰蒂肥大、類似男性的尿道下裂,大陰唇似男孩的陰囊,但無睪丸,或有不同程度的陰唇融合。雖然外生殖器有兩性畸形,但內生殖器仍為女性型,有卵巢、輸卵管、子宮、患兒2—3歲後可出現陰毛、腋毛,於青春期,女性性徵缺乏,無乳房發育和月經來潮。
男孩表現為假性性早熟,出生時無症狀,生後4個月以後出現性早熟徵象,一般1—2後外生殖器明顯增大,陰囊增大,但睪丸大小與年齡相稱。可早期出現陰毛、腋毛、鬍鬚、痤瘡、喉結,聲音低沉和肌肉發達
男孩還是女孩運出現體格發育過塊,骨齡超出年齡,因骨骺融合早,其最終身材矮小,由於ACTH增高,可有皮膚黏膜色素沉著。
(2)失鹽型(SW)
是21—羥化酶完全缺乏所致,皮質醇的前體物質如孕酮、17—羥孕酮等分泌增多,而醛固酮合成減少,使遠端腎小管排鈉過多,排鉀過少,因為,患兒除具有上述男性化表現外,生後不久即可有拒食、嘔吐、腹瀉、體重不增加或者下降、脫水、低血鈉、高血鉀、代謝性酸中毒等。若治療不及時,可因循環衰竭而死亡。女性患兒出生時已有兩性畸形,易於真噸,男性患兒診斷較為困難,常誤診為幽門狹窄而手術或誤診為嬰兒腹瀉而耽誤治療。
(3)非典型型(NC)
亦稱遲髮型、隱匿型或輕型,是由於21—羥化酶輕微缺乏所致。本症的臨床表現各異,發病年齡不一。在兒童期或青春期才出現男性化表現。男孩為陰毛、性早熟、生長加速、骨齡提前;女性患兒可出現初潮延遲、原發性閉經、多毛症及不育症等。
11β—羥化酶缺陷症(11β—OHD)
約占本病的5%—8%,此酶缺乏時,雄激素和11—脫氧皮質酮均增多,臨床表現出與21—羥化酶缺乏相似的男性化症狀,但程度較輕,可有高血壓和鈉瀦留 ,多數患兒血壓中等程度增高,其特點是給予糖皮質激素後血壓可下降,而停藥後血壓又回升。
3β—羥類固醇脫氫酶缺乏症(3β—HSD)
本型較罕見,該酶缺乏時,醛固酮 、皮質醇 、睪丸酮 的合成均受阻,男孩出現假兩性畸形,如陰莖發育差、尿道下裂。女孩出生時出現輕度男性化現象。由於醛固酮分泌低下,在新生兒期即發生失鹽、脫水症狀,病情較重。
17—羥化酶缺乏症(17—OHD)
本型亦罕見,由於皮質醇和性激素合成受阻,而11—脫氧皮質酮分泌增加,臨床出現低鉀性鹼中毒和高血壓,由於性激素缺乏,女孩可有幼稚型性徵、原發性閉經等,男孩則表現為男性假兩性畸形,外生殖器女性化,有乳房發育、但患兒有睪丸。
檢查診斷
生化檢測
 先天性腎上腺皮質增生症影響因素
先天性腎上腺皮質增生症影響因素 1、尿液17—羥類固醇(17—OHCS)、17—酮類固醇(17—KS)和孕三醇測定,其中17—KS是反映腎上腺皮質分泌雄激素的重要指標,對本病的診斷價值優於17—OHCS。腎上腺皮質增生症患者17—KS明顯升高。
2、血液17—羥孕酮(17—OHP)、腎素血管緊張素原(PRA)、醛固酮(Aldo)、脫氫異雄酮(DHEA)、脫氧皮質酮(DOC)及睪酮(T)等的測定,17—OHP基礎值升高是21—羥化酶缺乏的特異性指標,它還可用於檢測藥物劑量和療效。
3、血電解質測定:失鹽型可有低鈉、高甲血症。
其他檢查
1、染色體檢查
外生殖器 嚴重畸形時,可做染色體 核型分析,以鑑別性別。2、X線檢查
拍攝左手腕張支骨正位片,判斷骨齡,患者骨齡超過年齡。
3、B超或CT檢查
可發現雙側腎上腺增大。
4、基因診斷
採用直接聚合酶鏈反應,寡核苷酸 雜交,限制性內酶片段長度多態性和基因訊序列分析可發現相關基因突變或缺失。
治療
 先天性腎上腺皮質增生症的藥物治療
先天性腎上腺皮質增生症的藥物治療治療本病的目的:①糾正腎上腺皮質激素缺乏,維持正常生理代謝,②抑制男性化,促進正常的生長發育。
該病是完全可以治癒的,主要是藥物治療。女孩陰蒂增大,需手術治療,手術最適宜年齡為6個月~1歲。經過治療的患兒應根據年齡和開始治療後的反應,需要每3~12個月複查一次,以調整藥物的用量。
糾正治療
及時糾正水、電解質紊亂(針對失鹽型患兒):靜脈補液可用生理鹽水,有代謝性酸中毒則用0.45%氯化鈉和碳酸氫鈉溶液。忌用含鉀溶液。重症失鹽型需靜脈滴注氫化可的松25—100mg,若低鈉和脫水不易糾正,則可肌肉注射醋酸脫氧皮質酮(DOCA)1—3mg/d或口服氟氫可的松0.05—0.1mg/d,脫水糾正後,糖皮質激素改為口服,並長期維持,同時口服氯化鈉2—4d/d。其量可根據病情適當調整。
長期治療
(1)糖皮質激素
糖皮質激素 治療一方面可補償腎上腺分泌皮質醇的不足,一方面可抑制過多的ACTH釋放,從而減輕雄激素的過度產生,故可改善男性化、性早熟 等症狀,保證患兒正常的生長發育過程。一般氫化可的松 口服量為每日10—20mg/m2,2/3量睡前服,1/3量早晨服。
(2)鹽皮質激素
鹽皮質激素可協同糖皮質激素的作用,使ACTH的分泌進一步減少。可口服氟氫可的松0.05—0.1mg/d,症狀改善後,逐漸減量,停藥。因長期套用可引起高血壓。0.1mg氟氫可的松 相當於1.5mg氫化可的松,應將其量計算於皮質醇的用量中,以免皮質醇過量。
在皮質激素治療的過程中,應注意監測血17—羥孕酮或尿17—酮類固醇,失鹽型還應該監測血鉀、鈉、氯等。調節激素用量,患兒在應激情況下(如:感染、過度勞累、手術 等)或青春期 ,糖皮質激素的劑量應比平時增加1.5—2倍。
手術治療
男性患兒勿需手術治療。女性兩性畸形患兒宜6個月—1歲陰蒂部分切除術或矯形術。
疾病預防
新生兒篩查
運用乾血滴紙片法,經ELSA、螢光免疫法測定17—OHP可篩查21—OHD。產前診斷
(1)21—OHD:在孕9—11周取絨毛膜活檢進行胎兒細胞DNA分析,孕16—20周取羊水檢測孕三醇,17—OHP等,因大部分非典型21—OHD患兒生後17—OHP水平無明顯升高,因而基因檢測是此型患兒童唯一早期診斷手段。(2)11β—OHD:主要測羊水DOC及取絨毛膜作相關基因分析進行診斷。

