
病因
 肝豆狀核變性
肝豆狀核變性臨床表現
臨床主要表現神經精神症狀與肝臟症狀兩大方面。1.神經精神症狀
(1)震顫早期常限於上肢,漸延及全身。多表現為快速、節律性、似撲翼樣震顫,可並有運動時加重的意向性震顫。
(2)發音障礙與吞咽困難多見於兒童期發病的HLD說話緩慢似吟詩,或音調平坦似念經,也可有含糊不清、暴發性或震顫性語言。吞咽困難多發生於晚期患者。
(3)肌張力改變大多數患者肌張力呈齒輪樣、鉛管樣增高,往往引致動作遲緩、面部表情減少、寫字困難、步行障礙等。少數舞蹈型患者伴肌張力減退。
(4)癲癇發作較少見。
(5)精神症狀早期病人智慧型多無明顯變化,但急性起病的兒童較早發生智力減退;大多數肝豆狀核變性具有性格改變,如自制力減退、情緒不穩、易激動等;重症可出現抑鬱、狂躁、幻覺、妄想、衝動等,可引起傷人自傷行為。少數患者以精神症狀為首發症狀,易被誤診為精神分裂症。
2.肝臟症狀
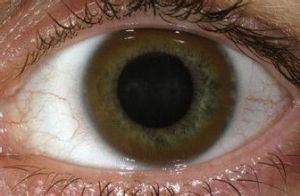 肝豆狀核變性 (棕色K-F環)
肝豆狀核變性 (棕色K-F環)(2)半數患者在5~10歲內出現一過性黃疸、短期谷丙轉氨酶增高或/及輕度腹水,不久迅速恢復。數年後當神經症狀出現時,肝臟可輕度腫大或不能捫及,肝功能輕度損害或正常範圍,但B超檢查已有不同程度損害。
(3)少兒期緩慢進行食欲不振、輕度黃疸、肝大和腹腔積液,酷似肝硬化的表現。經數月至數年,消化道症狀遷延不愈或日益加重,而漸漸出現震顫、肌僵直等神經症狀。神經症狀一旦出現,肝症狀迅速惡化,多於幾周至2~3個月內陷入肝昏迷。因此,對原因不明的肝硬化患兒應排除本病。
(4)部分青少年患者可表現緩慢進行脾臟腫大,並引致貧血、白細胞或(及)血小板減少等脾功能亢進徵象,一般在脾切除或/及門脈分流術後不久出現神經症狀並迅速惡化,常於短期內死亡;少數患者因食管靜脈破裂致上消化道出血而迅速促發神經症狀。
3.角膜色素環
肉眼或裂隙燈在角膜後彈力層周邊部可見棕色K-F環。
檢查
1.銅含量測定(1)頭髮銅含量測定對肝豆狀核變性診斷及鑑別診斷的價值不大。
(2)肌肉合銅量測定部分診斷困難的擬診肝豆狀核變性患者有一定的參考價值。
(3)皮膚和離體皮膚培養成纖維細胞內合銅量測定肝豆狀核變性患者明顯高於正常對照者(3倍)。
(4)指甲含銅量測定指甲含銅量測定是一種無損傷性的檢查方法,其優缺點與頭髮銅測定相同。
(5)膽汁內含銅量測定對肝豆狀核變性的診斷有特異價值。肝豆狀核變性患者膽汁內含銅量顯著減低。
2.影像學檢查
(1)肝豆狀核變性的肝臟B超檢查有其特殊的聲像圖,並將肝實質的聲像圖按肝損害的不同程度依次分為光點閃爍型、岩層征型、樹枝狀光帶型和結節型,對肝豆狀核變性具有特徵性診斷價值。對尚未出現神經症狀的肝豆狀核變性肝硬化者(結節型)與慢性肝炎肝硬化者有鑑別價值。可評估脾臟大小、形態。可顯示膽結石、腎結石、腎鈣質沉著。
(2)食管鋇劑造影攝片脾門靜脈造影或動脈造影可對疑有門脈高壓臨床表現的肝豆狀核變性患者進一步確診,有助於治療方案的制訂。
(3)骨關節X線檢查在肝豆狀核變性診斷上的意義:①骨關節X線改變是本病潛在的診斷指標。臨床上難以確診病例,不管有無骨關節症狀,都可利用該檢查幫助診斷。②在兒童、少年期出現不明原因的病理性骨折或X線照片發現腕、膝關節異常,要考慮到患肝豆狀核變性的可能性。③通過先證者做家系調查時可做為判斷是否為症狀前或症狀早期患者的輔助方法。
(3)顱腦CT、MRI無症狀的肝豆狀核變性及無腦症狀的肝型肝豆狀核變性患者顱腦CT掃描以腦萎縮為多見,而腦型肝豆狀核變性則以基底節區對稱性低密度影為特徵。因此,CT掃描對不典型的潛伏型、肝型及腦型肝豆狀核變性患者都有輔助診斷價值,但肝豆狀核變性的CT改變無特異性。肝豆狀核變性腦部MRI檢查,可顯示出比CT更為清晰的顱內異常表現,臨床意義與CT掃描相似。侵犯基底節神經核團時均表現為雙側對稱性,且為豆狀核、尾狀核頭部的大部分受累,而丘腦則為局部受累。腦幹病灶則以腦橋和中腦病變為主,少見小腦病灶。因而,對稱性基底節異常信號同時伴有腦幹病灶是肝豆狀核變性的影像特徵之一。
3.電生理檢查
(1)腦電圖以腦症狀為主的腦型肝豆狀核變性患者腦電圖多正常或輕度異常;以肝臟損害為主的腹型或肝型肝豆狀核變性患者的腦電圖多為中度、重度異常。腦電圖檢查有助於對有癲癇發作的肝豆狀核變性進行診斷。
(2)腦幹聽覺誘發電位(BAEP)肝豆狀核變性患者可出現BAEP異常,有一定的輔助診斷價值。
4.心理測試及IQ檢測
對精神障礙型肝豆狀核變性或呈現精神症狀的其他類型肝豆狀核變性,可通過心理測試以區別屬於行為障礙或器質性精神病。IQ檢測能了解患者智慧型障礙的程度。
5.其他檢查
(1)Tc膠態硫同位素掃描可清晰地顯示肝、脾的大小及形態。
(2)腹腔鏡檢查可看到肝臟硬化結節,有助於直接了解肝豆狀核變性患者肝臟損害的程度。
診斷
1.家族遺傳史,父母是近親婚配、同胞有肝豆狀核變性患者或死於原因不明的肝病者。2.緩慢進行性震顫、肌僵直、構語障礙等錐體外系症狀、體徵或/及肝症狀。
3.肉眼或裂隙燈證實有K-F環。
4.血清銅藍蛋白(CP)<200mg/L或血清銅氧化酶<0.2活力單位;血清總銅量低於正常值的1/2以下(4.7~14.1μmol/L)。
5.肝銅>250μg/g(乾重)。
判斷:①凡完全具備上述1~3項或2及4項者,可確診為臨床顯性型。②僅具有上述3~5項或3~4項者屬無症狀型肝豆狀核變性。③僅有1、2項或1、3項者,應懷疑肝豆狀核變性。
鑑別診斷
1.肝型肝豆狀核變性需與慢性活動性肝炎、慢性膽汁瘀滯綜合徵或門脈性肝硬化等肝病鑑別。但肝病無血清銅減低、尿銅增高、血清銅藍蛋白和銅氧化酶顯著降低等銅代謝異常,亦無角膜K-F環。2.假性硬化型肝豆狀核變性需與帕金森病鑑別,肝豆狀核變性型肝豆狀核變性需與特發性肌張力障礙鑑別。但帕金森病、特發性肌張力障礙均無銅代謝異常及角膜K-F環,可與肝豆狀核變性區別。
併發症
肝豆狀核變性患者免疫功能部分低下,部分患者有假性延髓麻痹的症狀,如吞咽困難、飲水反嗆等,特別是長期臥床的病人更容易患墜積性肺炎、尿路感染與褥瘡。有錐體外系症狀的患者,行走困難、易跌倒而出現骨折。肝豆狀核變性患者在肝硬化失代償期有門靜脈高壓合併食管-胃底靜脈曲張者,易出現急性上消化道出血,甚至發生出血性休克;少數肝臟的解毒能力下降,易出現肝性腦病、肝腎綜合徵等;亦有患者由於腦部損害而合併癲癇發作。
治療
1.低銅飲食每日食物中含銅量不應>1mg,不宜進食動物內臟、魚蝦海鮮和堅果等含銅量高的食物。
2.藥物治療
可套用二巰基丙醇、二巰丁二酸、二巰丙磺酸鈉、D-青黴胺、依地酸二鈉鈣、及鋅製劑等藥物。

