
疾病介紹
發作性睡病:又發作型睡病
 發作性睡病
發作性睡病白天過度嗜睡是發作性睡病首先出現的症狀,患者表現為突然出現無法預計的過度睡意和無法抗拒的睡眠發作。這種情形常出現在不適宜的場合,例如在吃飯、開車及工作中突然睡著。常可見到此類患者有數度交通意外或工作意外的經驗,患者有時會因此失去工作。但發作性睡病的患者卻經常抱怨晚上失眠,可能因頻繁的入眠期幻覺或睡眠癱瘓所導致晚上睡眠斷斷續續,無法一覺到天亮;也可能是因為白天嗜睡的時間過長,而干擾到晚上的睡眠。發作性睡病,一天24小時的總睡眠時數,並不比正常人長。
發作性睡病是以不可抗拒的短期睡眠發作為特點的一種疾病。多於兒童或青年期起病,男女發病率相似。部分病人可有腦炎或顱腦外傷史。其發病機制尚未清楚,可能與腦幹網狀結構上行激活系統功能降低或橋腦尾側網狀核功能亢進有關。多數病人伴有猝倒症、睡眠麻痹、睡眠幻覺等其他症狀,合稱為發作性睡病四聯症。
發作性睡病(narcolepsy)一詞,由Gelineau於1880年首創,用來描述在短時間內不自主的短暫的反覆睡眠為特徵的一種病理狀況,所以此病又稱Gelineau綜合徵。
直到1975年第一屆發作性睡病國際研討會,才對發作性睡病做如下定義:發作性睡病是一種病因不明的綜合徵,以具有發生異常睡眠的傾向為特徵,包括白天嗜睡和經常夜間睡眠異常,出現病理性快動眼睡眠表現。快動眼睡眠異常,包括睡眠一開始就進入快速動眼睡眠期,與快動眼抑制無關的猝倒和睡眠癱瘓。
分類
 Narcolepsy
Narcolepsy發作性睡病屬於睡眠障礙中“非呼吸相關的睡眠障礙所致白日過度睡眠”的一種,睡眠障礙國際分類第二版(ICSD-2)中將其分為4種亞型:
(1)發作性睡病,伴猝倒症;
(2)發作性睡病,不伴猝倒症;
(3)發作性睡病,醫學狀況所致;
(4)發作性睡病(待分類)。
原因
發作型睡病是指以白天出現陣發性不可抗拒的睡眠為特徵的睡眠障礙。據美國史丹福大學發作性睡病研究中心統計,美國人群的患病率大於0.05%。西歐國家人群的患病率約為0.02%~0.06%,日本人較常見,患病率為0.16%~0.18%。香港通過問卷調查及睡眠監測發現中國人的患病率約為0.034%。國外報導該病患病率約為0.2~0.9%,男女患病率無明顯差異,多數病例始發於10歲以後,10歲以前發病者約占5%。病因不明,可能與遺傳因素及睡眠機制異常有關。
其病因目前未明,但可發現此病跟基因、環境因素及某些中樞神經疾病有關。對可能誘發發作性睡病的環境因素現在知之甚少,文獻報導有包括頭部外傷、睡眠習慣改變、精神刺激及病毒感染等。而發作性睡病與遺傳基因的關係是近年來的研究熱點,大約8%~10%的發作性睡病患者具有家族史,患者直系親屬患病的幾率為對照組的10~40倍;25%~31%的單卵雙生子共患發作性睡病,提示遺傳因素在其起病中有重要作用。
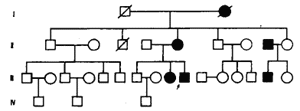
《中華醫學遺傳學雜誌》2000年第二期曾報導一例發作性睡病的家系圖譜
 發作性睡病
發作性睡病該家系4代31人,有6人發病,其中男3例,女3例,均有白天不可抗拒的睡眠。起病年齡分別為先證者(箭頭表示)的姐姐(三代-9)16歲,母親(二代-5)15歲,舅父(二代-8)19歲,表哥(三代-15)17歲,外祖母(一代-2)17歲,其中先證者母親、外祖母伴有猝倒發作。該家系連續3代均有人發病,其遺傳方法符合常染色體顯性遺傳。
根據研究,發作性睡病的病人體內常可偵測到HLA-DR2這個基因,但我們不能單單以HLA-DR2來判斷是否有發作性睡病,因正常人也有30%的人有HLA-DR2。並且有些疾病亦會造成HLA-DR2呈陽性反應。如果你的親人中,有人有發作性疾病,那您患發作性睡病的機率, 將是其他沒有家族史的人的6到18倍。
特徵
主要表現為短暫性睡眠發作、猝倒、入睡前幻覺及睡眠性癱瘓,合稱為發作型睡病四聯症。此四聯症並非每
 發作性睡病
發作性睡病1.白天過度嗜睡 主要表現為白天有不可抗拒的短暫睡眠發作,常在起床後3~4小時發生,發作時雖力求保持清醒,但不能自制,很快即進入睡眠狀態,睡眠一般持續數分鐘,每日可發作多次。發作不擇時間、地點及活動情況。雖然睡眠發作常在環境環境單調令正常人也會入睡的情況下發生,但也可以發生在具有危險性的情況下(例如駕車、橫穿馬路、高空危險作業等進行的途中)。醒後感到精力充沛、頭腦清楚,如果阻止其入睡則煩躁易怒。可以醒後數分鐘重又突然入睡。
雖然白天有頻繁的睡眠發作,但患者一天總的睡眠時間通常未見增加,在腦電圖記錄中可以觀察到發病一開始就立即進入快眼動睡眠期(REM),而正常的快眼動睡眠出現之前先有非快眼動睡眠,且通常持續60~90分鐘。而患者的夜間睡眠往往不能令人滿意,可被生動而可怕的夢境打斷。
2.猝倒 表現為突然出現的不自主低頭或突然倒地,但意識始終清楚,通常只持續幾秒鐘,一般每天只發作一次。猝倒是由於一過性部分或全身肌張力完全喪失引起的。
在65%-70%發作性睡病患者中可見到猝倒現象,猝倒是發作性睡病的特徵性表現之一。通常因有情緒上的刺激,如大笑、憤怒、興奮等所誘發。病人會突然膝蓋無力而跌倒; 或頭部突然失去肌肉張力而向後仰或向前低頭; 或突然面部肌肉張力喪失而導致面無表情,講話模糊不清。這三種臨床表現,是最常見到的猝倒症狀。發作時間通常少於一分鐘,而且意識清醒,無記憶障礙、呼吸完好,恢復完全。這些發作症狀與快眼動睡眠中發生的肌張力喪失,或在較輕的程度上與正常人在“笑得混身無力”時的情況相似。
3.入睡前幻覺 表現為入睡前或覺醒前的生動的夢樣體驗,以幻聽最為常見,亦可見幻視和幻觸。
入睡前幻覺和醒後幻覺見於12%-5
 發作性睡病
發作性睡病0%的發作性睡病患者,患者可在入睡前或覺醒時出現生動的、常常是不愉快的感覺性體驗,包括視覺、觸覺、運動或聽覺等。可表現為像做夢一樣的經歷,如看見體育場上運動員、走動的人等,常見的幻覺性體驗為身處火災現場、被人襲擊或在空中飛行等。這些幻覺非常生動,多是覺醒和睡眠轉換時出現的噩夢,偶伴有全身麻痹、壓迫感和恐懼感。患者常常敘述這些幻覺比一般的夢更可怕,因為這種夢境是從真實的(醒著的)環境中而來,區分現實狀態與夢境十分困難。
4.睡眠性癱瘓 發生在似睡非睡時,表現為不能動彈,也不能發聲。一般只持續幾秒鐘,偶而可長達十幾分鐘,他人呼喚和推搖,可中止發作。
睡眠癱瘓又叫睡眠麻痹,見於15%-34%的發作性睡病患者。睡眠癱瘓是發作性睡病患者從睡夢中醒來時發生的一過性的全身不能活動或不能講話,僅呼吸和眼球運動不受影響的恐怖體驗。睡眠癱瘓可以持續數秒到數分鐘,常與睡前幻覺和醒後幻覺同時發生,因此這種恐懼的感覺體驗得到了強化。睡眠癱瘓與快眼動睡眠中伴發的運動抑制很相似,在正常兒童及某些其他各方面都正常的成人中也屬常見。
不到半數的發作性睡病患者可出現上述所有4種症狀,約有85%的病人在發作性睡病病發前,有一些明顯的誘發因子存在著,例如嚴重的睡眠不足,
睡眠覺醒周期非常不規則,長久的晝夜輪班工作及頭部傷害(例如頭部外傷、腦瘤及多發性硬化症)等。此病的發病率約萬分之三到萬分之十六,每個年齡層均可能發生,尤好發於十來歲的青少年,並且男性比例稍多於女性。
體格檢查無神經系統陽性體徵。
治療
1.支持性心理治療 向患兒家長、老師及本人解釋該病的性質,協助其合理安排時間,允許其上課時小睡片刻。避免參加各種危險活動。
2.中醫治療 發作性睡病是由中氣不運所引起的,中氣即是脾胃之氣,祖國醫學有脾困人則困之說。治療發作性睡病是以補中益氣,健脾醒腦的治療原理採用中藥組方治療,此方有利於下丘腦分泌素的分泌,可使人體恢復清醒狀態,從而以此來治療發作性睡病。
目前有許多種有效且副作用小的藥物, 可用來控制發作性睡病的四大主要症狀。另外根據個別患者調整進食、小睡及運動的時間,可設計符合患者所需的行為治療,可幫助患者用最小的劑量,達到最大的效果。目前只能控制, 無法治癒。
檢查
根據短暫發 發作性睡病
發作性睡病作性不可抗拒的睡眠或伴有猝倒、睡眠麻痹、睡眠幻覺等典型症狀,一般診斷不難。但須與下列疾病鑑別。
一、癲癇失神發作。多見於兒童或少年,以意識障礙為主要症狀,常突然意識喪失,瞪目直視,呆立不動,並不跌倒;或突然終止正在進行的動作,如持物落地,不能繼續原有動作,歷時數秒。腦電圖可有3Hz的棘-慢綜合波。
二、昏倒。由於腦血液循環障礙所致短暫的一過性意識喪失。多有頭昏、無力、噁心、眼前發黑等短暫先兆,繼之意識喪失而昏倒。常伴有植物神經症狀,如面色蒼白、出冷汗、脈快微弱、血壓降低,多持續幾分鐘。
三、Kleine-Levin綜合徵。又稱周期性嗜睡與病理性飢餓綜合症。通常見於男性少年,呈周期性發作(間隔數周或數月),每次持續3~10天,表現為嗜睡、貪食和行為異常。病因及發病機制尚不清楚,可能為間腦特別是丘腦下部功能異常或局灶性腦炎所致。 是一種發作性睡眠過度,發作時僅在進食與大小便時才醒來,進食量可多至正常飲食的三倍以上,伴有激惹、躁動不安等。發作間歇期正常,常在成年後自然痊癒。
輔助檢查:
1、腦電圖。發作時腦電圖上很快出現快眼動睡眠中典型的低電位快活動。
2、腦脊液細胞檢查。腦脊液中下丘腦分泌素水平明顯降低。
3、CT和腦活檢。常規的臨床檢查如頭顱CT及MRI多難有異常發現。
4.多導生理記錄儀睡眠呼吸監測(PSG)及多次小睡睡眠潛伏期時間試驗(MSLT)可以發現睡眠潛伏期縮短及異常的快眼動睡眠,具有較大的診斷價值。
5.特定的人類白細胞抗原(HLA)基因測定。HLA-DR一般均為陽性。但此並不能成為診斷標準,因為有報導家族成員表現為和先證者相同的HLA-DR2組織單型但並不發病。
診斷
發作性睡病的診斷標準為:
(1)具有嗜睡及發作性猝倒的典型臨床表現;
或(2)具有白天嗜睡及以下表現者:平均睡眠潛伏期<8分鐘,2次或以上異常快眼動睡眠且無其他精神神經疾患及藥物能夠解釋嗜睡原因及有關症狀者。
少數患者只有睡眠發作一種症狀,也可能缺乏典型的快眼動睡眠的早期出現。

