流行病學
因子Ⅹ缺乏是一種罕見的疾病,在人群中的發病率低於1/50萬。在首例報導後已有超過50例的病例報導,但是,推測應該有更多的患者未被發現。病因
FⅩ是由肝臟合成的依賴維生素K的凝血因子。肝臟首先合成一條包括488個胺基酸組成的單鏈分子(包括由40個胺基酸殘基組成的信號肽)。FⅩ在凝血過程中被FⅨa/FⅧa或FⅦTF激活。一經激活,FⅩa就與它的必需輔因子(FⅤa)結合催化凝血酶原成為凝血酶當凝血因子Ⅹ缺乏時凝血酶的產生也相應延遲。發病機制
 遺傳性凝血因子Ⅹ缺乏
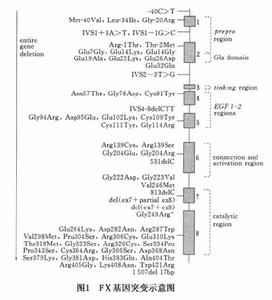
遺傳性凝血因子Ⅹ缺乏遺傳性凝血因子Ⅹ缺乏是常染色體隱性遺傳性疾病編碼FⅩ的基因位於13號染色體,已被成功克隆和測序,FⅩ基因長22kb,包含8個外顯子目前,已經發現60餘種突變,其中絕大多數是錯義突變,主要發生於編碼催化結構域的8號外顯子。所有這些突變都不會導致截短型蛋白的產生也不會消除FⅩ的表達,這就從另一個側面說明為什麼在FⅩ基因剔除小鼠中,FⅩ完全不表達的小鼠是不能夠存活下來的臨床中,大多數患者的活性雖然減低,但是,仍可被測出,抗原水平減低或正常,非常嚴重的突變如缺失或剪下位點突變所占比例極小。在FⅩ基因突變譜中非常有趣的一點是目前尚沒有發現有無義突變存在,而在其他的遺傳性凝血因子缺乏中,這種類型的突變約占所有突變的1/5。FⅩFruili的純合子有嚴重的出血表現FⅩ活性只有正常的6%~9%,但是,抗原水平卻是正常的。其他類似的家系也有報導。
上海瑞金醫院上海血液學研究所對一個FⅩ缺乏家系的先證者和其他成員FⅩ基因所有外顯子及其側翼內含子序列進行DNA測序,發現FⅩ基因外顯子1錯義突變11Set(AGT)→Arg(AGG)該突變為國際首次發現圖1所示為部分FⅩ基因突變和其在基因中的位置.
臨床表現
除血友病甲和血友病乙之外的凝血因子缺乏中,因子Ⅹ缺乏患者的臨床出血表現最為嚴重。在2/3的患者中可能發生血腫和關節出血,部分患者甚至可能出現胃腸道出血或致命性的臍帶殘端出血。其出血表現的輕重與凝血活性受抑的程度有關。當FⅩ活性低於1%時患者出現嚴重出血,當FⅩ水平≥10%可能僅表現為輕微出血FⅩ活性低於1%的患者臨床表現與血友病甲類似。
診斷
根據臨床出血症狀、遺傳類型和實驗室檢查進行診斷,FⅪ:C測定或Biggs凝血活酶生成試驗可確定診斷。
鑑別診斷:
1.本病主要與凝血酶原時間(PT)正常部分凝血活酶時間(PTT)延長的其他出血性疾病鑑別,Biggs凝血活酶生成試驗可與血友病甲和血友病乙鑑別。狼瘡抗凝物質可使PTT延長PT正常,狼瘡抗凝物質的實驗室檢查可資鑑別。獲得性FⅪ缺乏症的鑑別在於此類患者存在自身抗體,可用抗體篩選試驗鑑別,常發生於系統性紅斑狼瘡病例。
2.遺傳性凝血因子Ⅹ缺乏的診斷必須與繼發於維生素K缺乏的獲得性FⅩ減少相鑑別肝臟疾病和華法林也可以表現出因子Ⅹ缺乏的症狀,但是在上述兩種情況中,FⅩ的減少也是繼發性的並且同時還會有其他依賴維生素K的凝血因子缺乏,通過詳細的病史,體格檢查和實驗室檢查可以明確診斷。在澱粉樣變性的患者中可以發現孤立的獲得性凝血因子Ⅹ缺乏,其發生的原因可能與澱粉樣蛋白對FⅩ的吸收有關。
檢查
凝血酶原時間(PT)和活化部分凝血活酶時間(APTT)通常都延長,但是,由於FⅩ必須與FⅨa/FⅧa複合物和FⅦa/TF複合物相互作用,因此,當FⅩ發生缺乏時可能對兩種複合物作用影響並不相同。例如,在FⅩRoma中,FⅩ的抗原水平正常,但是,其對外源性凝血途徑的影響(3%)要比對內源性凝血途徑影響(30%~50%)大得多。具有這種突變的患者具有出血素質在其他病例中也可以發現僅PT延長,而APTT正常,或是APTT延長,而PT正常。嚴重缺乏FⅩ的患者出血時間也可能延長,但是,是否出血時間的延長與FⅤa和FⅩa在血小板表面相互作用的障礙有關尚不是十分清楚蝰蛇毒可以直接裂解和活化FⅩ,因此,Russell蝰蛇毒時間檢查在多數患者中延長。對FⅩ活性和抗原,以及基因學的檢查是明確遺傳性凝血因子Ⅹ缺乏的診斷所必需的。
治療
替代治療是本病出血症狀的主要治療方法。一般輕微出血不需要治療。外傷後嚴重出血,手術後出血均需替代治療。FⅪ半衰期約52h,因而隔天輸注1次即能維持血漿水平。FⅪ彌散率低,容易提高血漿水平國內尚無濃縮FⅪ製劑,可用新鮮血漿或新鮮冰凍血漿,也可用已去除冷沉澱的上清血漿。血庫全血在1周內損失約80%的FⅪ,因而不適用。一般每kg體重給予5~20ml血漿可使FⅪ水平上升到25%~50%,達到有效止血水平。外科手術正常止血所需確切FⅪ水平並不清楚,一般認為應達到或超過50%。手術前輸注30ml/kg新鮮血漿可達到此水平術後每天5ml/kg或隔天10ml/kg新鮮血漿直至傷口癒合。國外已有濃縮FⅪ製劑。治療併發症主要為肝炎及其他與輸血有關的病毒的傳染如愛滋病病毒等。
輸注血製品後產生FⅪ抑制物(同種抗體)的病例出血嚴重,血漿替代治療止血無效,用激活的凝血酶原複合物可能有效。
預後預防
預後:
1.本病一般出血輕微,因出血導致的死亡率很低。預後取決於病例出血的嚴重性和替代治療併發症,出血輕微者預後良好。
2.嚴重缺乏FⅩ的患者可有與血友病甲和血友病乙類似的慢性關節疾病。由於反覆輸注血漿和凝血因子濃縮製劑使一些患者感染上B型肝炎或C型肝炎,進而導致慢性肝臟疾病
3.套用的濃縮製劑存在誘發血栓形成和DIC的危險。
預防:
建立遺傳諮詢嚴格婚前檢查加強產前診斷,減少患兒的出生。

