
病因
確切病因不明,6%為常染色體顯性遺傳,94%為散發病例,其中25%為遺傳突變,其餘為體細胞突變,亦有人認為與病毒感染因素有關。
概述
視網膜母細胞瘤(retinoblastoma)這是一種起源於胚胎視網膜細胞的惡性腫瘤,具有家族性和遺傳性傾向。多發生於3歲以前,205雙眼受累,個別的病例可發生在成年,甚至老年。發病率在眼部腫瘤中占據首位,占眼部腫瘤的33.8%,眼內腫瘤為70%。由於此瘤惡性程度高,並可引起全身轉移而招死亡,發現與治療較晚者,風險因素較大。故早期診斷十分重要,只要診斷及時,處理得當,治癒率可達50%以上,兒童腫瘤治療尤其謹慎,通過質子治療兒童腫瘤得到很好的效果,不損害周圍組織還能有效殺死癌細胞,最大的保護正常組織,通過攜康長榮國際轉診治療兒童腫瘤也是一個不錯的選擇。
發病原因
視網膜母細胞瘤的病因不明,個別遺傳視網膜母細胞瘤發生的危險因素有高齡父母(Derkinderen etal.,1990)及父親受僱於金屬工廠(Bunin etal.,1990),約1/10單側視網膜母細胞瘤患者攜帶視網膜母細胞瘤易感基因,故其下一代發生單側病變的危險為1/20。
發病機制
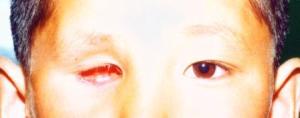 小兒視網膜母細胞瘤
小兒視網膜母細胞瘤雖然視網膜母細胞瘤的組織來源仍有爭論,近年的研究認為該瘤源於神經上皮,可被分類為原始神經外胚葉腫瘤(primitive neuroectdermal tumors,PNETs),腫瘤常發生於視網膜的後部分,瘤細胞小而圓,胞漿很少,密集,常形成玫瑰花結。
Reese-Ellsworth的腫瘤分期如下:
Ⅰ期:預後很好,單發或多發,腫瘤小於4盤直徑(disk diameter)(盤直徑=1.5~1.75mm)位於或在中緯線之後。
Ⅱ期:預後好,單發或多發腫瘤,4~10盤直徑,位於或在中緯線之後。
Ⅲ期:預後不定,病變位於中緯線之前;單發腫瘤大於10盤直徑,位於中緯線之後。
Ⅳ期:預後差,多發腫瘤,有些大於10盤直徑;任何病變擴散到視網膜鋸齒線。
Ⅴ期:預後很差,巨大腫瘤侵犯視網膜一半以上;玻璃體種植。
巨大腫瘤如擴散到脈絡膜可致血行播散,如腫瘤穿透篩板,沿視神經可侵犯中樞神經,由於這些腫瘤罕見轉移,因原發瘤就診時常可保存有效視力。
臨床表現
按腫瘤發展過程,常分為四期:①眼內生長期:②青光眼期;③眼外蔓延期;④轉移期。
實際上,人為的分期,只表明腫瘤的一般發展過程。然而,腫瘤不可能盡按上述順序發展。可能腫瘤很小,還無眼內壓增高已經轉移,也可發展到一定程度後又自行萎縮。一般說來,早期多從眼底之後極下方開始,起源於核心層者多見,常為許多灰白色小結節,或較大的白色結節被許多衛星小結節所包圍。以後的發展和腫瘤的起源有關,起源於核心層者從視網膜內面有腫塊向玻璃體突出,表面常有粗大新生血管或摻雜出血斑。扁平型者沿視網膜平面發展,視網膜血管粗大彎曲,可擬似視網膜血管瘤。不管腫瘤以何種形成開始,增大到一定程度後終將玻璃體腔填滿,顯示黃光反射,狀如貓眼,故有黑蒙性貓眼之稱。
1.眼內生長期:因腫瘤發生部位而異,假如腫瘤從前部開始,而且增長緩慢,可要相當長時期內保留一定視力。反之,從眼後極部開始的腫瘤,往往早期引起視力減退而導致眼球偏斜,故對小兒眼球偏斜不忽視與輕視。如果視網膜完全脫離,或腫塊充滿眼球內,可致視功能喪失。眼底檢查,可見視網膜上有圓形結節隆起,呈黃白色,境界清晰,腫塊上有新血管或出血。
2.青光眼期:眼內腫塊增大並向前後發展,特別景物到脈絡膜及前房角,常使眼壓急劇增高,呈急性閉角膜型青光眼狀態。由於嬰幼兒眼球壁富於彈性,在高眼壓下形成所謂牛眼,鞏膜葡萄腫。高度膨大突出的眼球容易發生角膜潰瘍,晶體體向玻璃體或前房脫位。患兒眼痛、頭痛、哭鬧不安。
3.眼外蔓延期:此期腫瘤可沿視神經向球後發展。①腫瘤侵入視神經多是直接蔓延的結果;②少數病例,腫瘤細胞經秦氏血管環再經神經鞘,沿蛛網膜下腔及軟腦膜進進球後視神經,然後進入顱腔,侵入腦組織;③經過鞏膜導水管離開眼球,進入眼眶,在球後及眶內迅速增大,而引起眼球突出。亦可向前突破角膜鞏膜向外發展,成為一個很大的腫塊嵌頓於瞼裂中。
4.全身轉移:沿血液及淋巴向全身轉移。據統計受累器官中,腦及腦膜占第一位,顱肌次之,再次為淋巴結及長骨,腹部器官以肝最為多見。
併發症
引起繼發性青光眼,前房積血,眼內炎症,可合併其他畸形,染色體13的長臂丟失綜合徵,肛門閉鎖,會陰瘺和拇指發育不良,智力遲鈍,生長遲滯,齶裂和多指(趾)畸形等。
檢查
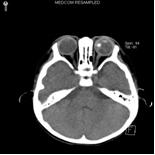 小兒視網膜母細胞瘤
小兒視網膜母細胞瘤幼兒瞳孔後有黃白色光反射,視力減退伴有斜視,原因不明的前房積血,單眼瞳孔擴大或青光眼,有視網膜母細胞瘤家族史者,則應深入檢查雙側眼底及玻璃 體,檢查宜於全麻下進行,散大瞳孔,用間接眼膜曲率鏡以查明眼內病變程度,鞏膜凹陷以及中緯線前的視網膜,對於晚期眼內腫瘤,尤以復發或有遠處轉移者應做 下述檢查:
1.腦脊液檢查 注意有無惡性腫瘤細胞。
2.血液檢查血清甲胎蛋白增高,手術切除腫瘤後血清甲胎蛋白可下降,腫瘤復發則再次升高。
3.骨髓檢查 注意檢查有無瘤細胞。
4.病理檢查 淋巴結腫大者可做淋巴結活檢。
1.眼底檢查雙側散大瞳孔仔細檢查眼底,雙眼的陽性發現有助於診治方案和判斷預後。
2.頭顱X線片檢查 可見鈣化斑,如視神經孔擴大,可考慮有顱內蔓延。
3.超音波檢查 超聲和CT檢查可顯示眼內及眼眶內的腫瘤病變。
A超在玻璃體平段內出現病理回聲不等的波峰,通常與眼球壁相連線,重度病例病理波峰可占據全部玻璃體腔,不同部位聲衰減不一致。
B超顯示眼球壁向玻璃體腔內出現邊界清楚,似球形或形狀不規則的回聲光團,內反射回聲光點強弱不等,分布不均,病變內有壞死,可出現囊性暗區圖像。
4.CT掃描檢查 CT檢查可顯示眼內及眼眶內的腫瘤病變,可見玻璃體腔內出現密度增高不均勻的局限性腫塊,常伴鈣化斑,晚期病例視神經增粗,視神經孔擴大。
5.眼底螢光血管造影檢查 是脈絡膜惡性黑色素瘤的一種有價值的輔助診斷方法。
鑑別診斷
不典型的病例常在臨床上被誤診為其它眼病,故應細緻的進行鑑別。
1.轉移性眼內炎:轉移性內眼炎發展到一定階段後,可因玻璃體膿腫的存在而在瞳孔中呈現黃色反射,足以混淆視網膜母細胞瘤的診斷。由於此二病在幼童中都是比較常見的眼病,兩者鑑別就更有必要。
⑴一則屬於急性化膿性炎症,總是或多或少地有一些炎症的表現。一則屬於惡性腫瘤,不僅病程較長,本質的表現也完全兩樣。例如:轉移性眼炎多繼於急性全身性感染之後,眼部常常有明顯的炎症反應,包括房水混濁及角膜後壁沉著物的出現、虹膜後粘連的形成及瞳孔變形、炎症引起的並發性白內障等。眼內腫瘤一般無此表現。
⑵腫瘤病例在發展過程中要引起繼發性青光眼,導致角膜或整個眼球的擴大,轉移性眼內炎則在不太長的時間內(約為半個到1個月)引起眼球萎縮。
⑶突出玻璃體內的瘤組織腫塊多有新生血管或出血,轉移性眼炎的黃光反射來自玻璃體的膿液,所以僅見黃光而缺乏出血或新生血管。
⑷腫瘤很少引起晶狀體混濁,眼內炎症並發白內障者卻甚多見。
⑸X線片上腫瘤病例多見有鈣化點存在,甚至在少數病例可有視神經孔的擴大,眼內炎患者則無此表現。視網膜母細胞瘤雖然常發生壞死,卻很少引起炎症。
2.視網膜母細胞瘤與寇次(Coats)病的鑑別診斷:寇次(Coats)病的根本性質是視網膜外層出血合併滲出性改變。雖有局限性增殖,甚至形成隆起或導致視網膜脫離。但病程緩慢,病變範圍較為廣泛,灰白色滲出物分布在視網膜血管之後。除滲出物外,還可見出血斑和光亮小點(膽固醇結晶體)沉著。血管尤其靜脈顯示擴張、扭轉、紆曲,並有微血管瘤。病變常為進行性,新舊滲出物可交替出現,出血如果進入玻璃體,可形成增殖性玻璃體視網膜病變。本病患者年齡較在,多在6歲以上,且為青年男性,單眼受累。超音波檢查,常無實質改變。
最佳治療方法
對單眼和雙眼母細胞瘤,應分別對對待。單側病例應及早摘除眼球,剪斷的視神經不應短於10mm。如果病理切片顯示視視斷端已受累,應考慮行眼眶內容摘除,輔以放療或化療。雙眼者,若一眼已失明,另一眼尚有視力,摘除視功能喪失的一眼,另一眼可行保守治療,採用光凝、冷凝、放療,雷射—血卟啉療法。雙眼均已失明,為挽救患者生命套用眼球摘除。
目前質子治療視網膜母細胞瘤的臨床。質子放療可以有效的避免射線對腦部主要功能區的照射,並且在入射皮膚時幾乎沒有劑量的損失,同時能在腫瘤部位實行大劑量爆破,能夠有效的殺死癌細胞。在歐美,兒童腫瘤的質子治療已經納入了保險,因為質子治療可以有效的保護眼球,這對於生長和發育的兒童來講是不二的選擇。
預防
應做好遺傳病諮詢工作,雙眼視網膜母細胞瘤患者多屬遺傳性,因此,對雙眼患者的子女和有家族史患者的子女,應高度警惕腫瘤的發生,以便早期發現,早期診斷和早期治療。
罕見病詞條庫

盤點常見的遺傳病
| 隨著分子生物學的發展,科學家們成功的把基因和疾病聯繫在一起。人們可以預先通過基因篩查來預測可能罹患的遺傳病。比如,GOOGLE公司創始人之一謝爾蓋?布林,就在其妻子的23andme生物技術公司通過測序發現自己可能在未來患帕金森氏綜合症,他積極通過各種方式來預防疾病的發生並投入巨資進行相關的研究。但如同一把雙刃劍,基因篩查也使一些攜帶突變基因的人在社會各個方面受到歧視,讓我們來關注一下遺傳疾病吧! |
| 黑棘皮症| 短趾症| 家族性高膽固醇血症| 白化病| 苯丙酮尿症| 紅綠色盲| 抗維生素D佝僂病| 先天性髖關節脫位| 脊柱裂| 先天性聾啞| 蠶豆病 | 強直性肌營養不良| 亨廷頓氏病| 老年性痴呆症| 精神分裂症| 脆性X綜合徵 | 原發性高血壓| WILSON 氏病| 成人多囊腎病| 馬凡綜合徵 | 侏儒綜合徵| 視網膜色素變性| 糖原貯積症| 神經鞘脂貯積症| 黏多糖貯積症 | 半乳糖血症| 白癜風| 性反轉綜合徵| WILMS 瘤| 視網膜母細胞瘤| 成骨不全病| 自毀容貌症| 家族性多發性結腸息肉病| 強直性脊柱炎| 假肥大性肌營養不良| 牛皮癬| 多囊腎| 脆骨病| 神經纖維瘤| 上瞼下垂| 類風濕性關節炎| 癲癇| 先天性心臟病| 高膽固醇血症| 唇裂| 齶裂| 魚鱗症| 多指| 著色性乾皮病| 腓肌萎縮症| 並指| 畸形足| 青光眼| 全身自化| 結腸息肉症| 先天聾啞| 原發性小睪症 |
