
簡介
 Marfan syndrome
Marfan syndrome馬凡綜合徵( Marfan syndrome)為一先天性中胚葉發育不良性疾病,為一遺傳型結締組織病,系常染色體顯性遺傳性疾病,個別呈常染色體隱性遺傳,具體發病原因不明,據認為與先天性蛋白質代謝異常有關。人群發病率約 4人/10萬人,本綜合徵臨床表現不一,主要累及骨骼、心血管系統和眼等器官組織。是法國兒科專家Antoine於1896首例報告,此後有類似病例報告,於1931年正式稱之Marfan綜合徵。
流行病學
 馬凡綜合徵
馬凡綜合徵本病有家族發病傾向,呈常染色體顯性遺傳。兩性發病,無種族差異,多見於兒童也可見於成人其發病情況沒有其他相關內容描述。
病因
 馬凡綜合徵
馬凡綜合徵本病呈常染色體顯性遺傳,在人體很多組織如心內膜心瓣膜、大血管、骨骼等處,均有硫酸軟骨素A或C等黏多糖堆積,從而影響了彈力纖維和其他結締組織纖維的結構和功能,使相應的器官發育不良及出現功能異常、Abraham等(1982)提出,主動脈彈性蛋白有異常,橋粒蛋白和異橋粒蛋白減少,而賴氨醯殘基相應增加,是該病的主要改變病人尿中羥脯氨酸排泄量有增高,血中黏蛋白和黏多糖也增高。
發病機制
 馬凡綜合徵
馬凡綜合徵通過家族的連鎖基因定位顯性遺傳,可以從病人尿中羥脯氨酸排泄量增多來證明,本病為彈力纖維缺損亦即膠原代謝異常。結締組織纖維是機體組織結構中很重要的成分,因此,當它發生異常時,便會影響全身的臟器(中胚層組織),尤以骨骼和心血管系統更為顯著。在蜘蛛指以及凹陷的胸部或舟狀胸部都表示了四肢管狀骨、手指和肋骨縱軸過度增長,可能是由於骨膜纖維成分缺陷的結果。在主動脈和肺動脈中層有酸性黏多糖沉積。本病有家族傾向,呈常染色體顯性遺傳。
病理
 馬凡綜合徵
馬凡綜合徵肉眼改變有眼的異常,升主動脈擴張慢性主動脈夾層瘤形成心臟瓣膜呈黏液性水腫性改變,瓣膜呈氣球形,腱索增粗心臟肥大和二尖瓣鈣化以及皮紋異常等。本綜合徵的病理改變以心血管系統最顯著並具有代表性。顯微鏡下可見有主動脈中層彈力組織稀疏和碎裂,伴平滑肌呈不規則的螺紋狀改變,膠原量增加,並可見有異染性物質呈囊狀空泡散在分布於中層主動脈夾層瘤形成者,顯示為囊狀中層壞死和彈力纖維中度變性,伴以平滑肌束紊亂。主動脈瓣的組織病理改變為正常結構破壞和喪失,囊狀變性和組織纖維細胞喪失。皮膚的病理改變表現為有空泡狀退行性變及彈力纖維排列紊亂。關節滑膜改變也是彈力纖維變性、膠原增多及異染性物質呈散在性囊狀分布。
臨床表現
 馬凡綜合徵
馬凡綜合徵兩性發病,無種族差異,多見於兒童,也可見於成人多數病人出生後即有症狀,面容顯老,表現為一種憂愁的外觀,軀幹纖細肌肉不發達,皮下脂肪菲薄。
骨骼改變
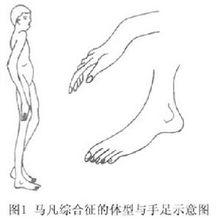
本綜合徵病人四肢奇長且細,尤以指(趾)為著。軀幹可因側彎後突而短縮,使四肢顯得更為伸長,宛如蜘蛛足,故名蜘蛛指(圖1)。肌肉張力降低,關節活動增加,可有超常的運動範圍,但脫位罕見。頭長,額部圓凸,胸骨畸形多由肋骨過長所致漏斗胸或雞胸更常見,肩胛隆起呈翼狀。全身性結締組織異常可累及關節囊、韌帶、肌腱、肌膜,可導致關節反覆脫位、扁平足或高弓足,齶弓高,牙齒不整齊。常見馬凡綜合徵檢查方法:
(1)掌骨指數:在雙手X線後前位片上,示指、中指、無名指和小指4個掌骨平均長度除以該4掌骨中部的平均寬度所得數值,正常人掌骨指數小於8,該綜合徵男大於8.4,女大於9.2。
(2)拇指征:令患者拇指內收,橫置於掌心伸直並握拳。如果伸展的拇指明顯超出該手尺側緣,則為陽性。
(3)腕征:患者以一手在對側橈骨莖頭近端處握住對側手腕,以拇指和小指圍繞1周如果拇指與小指不加壓力時可相互重疊則為陽性。
皮膚改變
最常見的皮膚表現為皮紋增寬或有萎縮性皮紋這些皮膚異常表現可見於身體的許多部位,尤以胸部、肩部三角肌區和大腿部為顯著。
心血管異常
30%~40%的病人有心血管系統併發症,最常見的心血管異常為主動脈特發性擴張、主動脈夾層動脈瘤和二尖瓣異常等。有時可同時發生主動脈病變和二尖瓣病變。伴有收縮晚期雜音的收縮期喀喇音是其最常見的體徵此外,外傷、高血壓和妊娠可以誘發急性主動脈破裂和夾層動脈瘤形成。除主動脈瓣和二尖瓣病變外,有時尚可發生三尖瓣病變。雖然主動脈擴張總是發生在升主動脈,但胸主動脈和腹主動脈也可發生動脈瘤樣擴張、夾層動脈瘤形成或破裂。約1/3的病人可並有先天性心臟病,常見為主動脈瓣狹窄、動脈導管未閉、房間隔缺損等其他少見的心血管併發症有佛氏竇和肺動脈擴張,主動脈的主要分支如頸總動脈、脾動脈擴張,心內膜纖維變性主動脈瘤破裂和心力衰竭是本綜合徵的主要死亡原因。
眼部改變
最特徵性表現是晶體脫位或半脫位,約3/4的患者為雙側性。晶體脫位可由多種因素所引起。大眼和小晶體可使晶體周圍間隙增大,懸韌帶擴展,睫狀體發育不良,懸韌帶及其附著於晶體處異常。此外,本綜合徵還可出現高度近視、青光眼、視網膜剝離、虹膜炎等眼部異常。這些眼部病變較晶體脫位對眼的影響更為嚴重。鞏膜異常表現為藍色鞏膜。有時也可發生角膜過大、色素性視網膜炎、脈絡膜硬化、斜視、眼球震顫、瞼震顫和前房變淺
神經系統病變
本綜合徵的神經系統症狀與其他先天性風濕病一樣,也是由腦血管畸形所造成,表現為蛛網膜下腔出血和頸內動脈瘤所致的壓迫症狀動脈瘤引起的癲癇大發作。此外,馬凡綜合徵病人還可發生脊柱裂脊柱脊髓膨出、脊髓空洞症。肌張力低下伴有肌萎縮是本綜合徵最常見的神經肌肉症狀。少數病人可有智力落後或痴呆。
併發症
1.心血管最可能並發,主動脈特發性擴張、主動脈瓣狹窄,主動脈夾層動脈瘤和二尖瓣異常等。
2.眼部病變可並發晶體脫位或半脫位,高度近視、青光眼、視網膜剝離、虹膜炎等
3.神經系統病變可並發,蛛網膜下腔出血和頸內動脈瘤,癲癇大發作。此外,馬凡綜合徵病人還可發生脊柱裂脊柱脊髓膨出、脊髓空洞症。
實驗室檢查
1. 裂隙燈檢查,可確定有無晶狀體異位。
2. X線檢查:指骨細長,掌骨指數≥8.4,(即右第 2-5掌骨長寬之比),正常為5.5~8.0。
3. 超聲心動圖:可見主動脈根部擴張、主動脈瓣關閉不全和其他並發的心臟畸形。
4. CT、核磁共振。較超聲心動圖更精確。
診斷
1.本綜合徵的診斷依據為
(1)特殊骨骼變化即管狀骨細長尤以指、掌骨為著。骨皮質變薄、纖細,呈蜘蛛指樣改變。
(2)先天性心血管異常
(3)眼部症狀。
(4)家族史。
以上臨床4項標準中有3項者即可確診,在前3項中僅出現2項改變即可診斷為不完全型馬凡綜合徵
2.Mckusick(1995)將馬凡綜合徵的心血管異常列為
(1)主動脈擴張(升主動脈、降主動脈),主動脈夾層瘤,主動脈瓣狹窄,動脈導管未閉。
(2)肺動脈異常(肺動脈擴張、肺動脈瘤)。
(3)間隔缺損(房間隔缺損、室間隔缺損)。
(4)瓣膜異常和伴有亞急性細菌性心內膜炎。
鑑別診斷:
本病需注意與下列疾病鑑別:
1.艾-盪綜合徵 雖可有四肢過長、關節過動症狀,但馬凡綜合徵不出現皮膚和血管脆弱及皮膚過度伸展症狀。
2.彈性假黃瘤病 可發生主動脈瘤和鬆弛性皮損皮損為丘疹或斑丘疹,邊界清楚呈點狀、圓形或橢圓形或融合成片,局部高起另外,該病有特徵性視網膜血管樣色素紋理。無關節過動也無肢端骨細長和蜘蛛指。
3.同型胱氨酸尿症 為一種先天性甲硫氨酸代謝異常疾病可有晶體脫位、肢端異常胸和脊柱異常。但尿的異常全身性骨質疏鬆、脈管栓塞和反應遲鈍等在馬凡綜合徵者不出現。
治療
無特殊療法,眼異常可進行相應的手術或藥物治療。主動脈病變時可服用普萘洛爾(心得安),使其心室排血和壓力減低,減輕主動脈壁承受的衝擊,因此,可延緩主動脈根部擴張的發展及防止主動脈夾層動脈瘤的發生對青春期前的女性患者,可服用雌激素及黃體酮以提前進入青春期,防止因生長過快造成脊柱側彎畸形嚴重胸廓、脊柱畸形患者、中度主動脈瓣閉鎖不全或主動脈根部明顯擴張患者,可採用手術治療。
預後預防
預後
較好,多數病人可存活到中年,常死於主動脈瘤破裂和心力衰竭。
預防
1.一級預防 遺傳病的預防除了從整個人群的角度做好流行病學調查攜帶者檢出、進行人群遺傳監護和環境監護,開展婚姻和生育指導,努力降低人群中遺傳病發生率提高人口素質之外,針對個體,必須採取有效的預防措施,避免遺傳病後代的出生(即實行優生)和遺傳變異的發生採取通常的措施包括:婚前檢查遺傳諮詢、產前檢查和遺傳病的早期治療。
(1)婚前檢查:婚前檢查(即婚姻保健),它是保證男女雙方婚後生活幸福、後代健康的重要環節。婚前檢查的重點是:①遺傳病方面的調查包括詳細詢問男女雙方及其家庭成員的健康狀況既往病史及醫治情況,尤其是有無先天畸形,遺傳病史和近親婚配史。必要時應進行家系調查、血型檢查染色體檢查或基因診斷,以檢出攜帶者;②全面的體格檢查,主要是對急性傳染病,結核病,或嚴重的心、肝腎疾病泌尿道慢性炎症等可嚴重威脅個人或配偶健康的疾病,以及女方的嚴重貧血、糖尿病等可對胎兒造成影響的疾病的檢出,並動員經治癒後才可結婚;③對男女生殖器官的檢查,檢出性器官畸形,兩性畸形等疾患,以便極早採取措施。
(2)遺傳諮詢:遺傳諮詢( genetic counselling)是由臨床醫生從遺傳學上,作肯定解答遺傳病患者及其親屬提出的有關遺傳性疾病的病因、遺傳方式、診斷、治療及預後等問題,估計患者的子女再患某病的機率,並提出建議及指導,以供患者及其親屬參考。遺傳諮詢的意義在於:①減輕患者身體和精神上的痛苦,減輕患者及其親屬的心理壓力幫助他們正確對待遺傳病、了解發病機率,採取正確的預防、治療措施;②降低人群遺傳病的發生率降低有害基因的頻率,及減少傳遞機會
①遺傳諮詢的分類和內容:
A.婚前諮詢:婚前男女雙方在得知一方或其親屬中有某種遺傳病時詢問能否結婚?後代中該病的發病情況怎樣?
B.產前諮詢:夫妻中的一方或其親屬中有某種遺傳病或先天性畸形,詢問後代類似疾病的發生情況;若已作過某種遺傳病或先天性畸形,詢問再生育時後代情況及如何預防患兒的出生。妊娠期曾患某病、服用某藥或接觸有毒物質或放射線,詢問胎兒的可能情況。
C.一般遺傳諮詢:除了上述的情況外,還詢問可否近親婚配?已經發病個體的防治辦法出現某些症狀或體徵疑為遺傳病者想得到解釋等。
儘管諮詢者的年齡職業、知識基礎和文化水平各不相同,來意和要求也不一樣,但遺傳諮詢的基本內容可以歸納為以下4方面:a.明確診斷是否屬於遺傳性疾病;b.解答各種問題,包括防治和預後;c.推算復發風險率;d.商討對策。為了完成這些內容,遺傳諮詢通常採用的程式包括:a.通過病史和家系調查,體格檢查和必要的輔助檢查和特殊的遺傳學檢查分析,確定是否為遺傳病,以何種方式遺傳;b.按照遺傳方式和特點估計復發風險率;c.通過商談討論、提出預防治療對策和婚姻、生育指導。
②復發風險率的估算:
A.人類遺傳病的復發風險,按其危險程度,可分為3類:
a.一般風險:發病機率為1∶20以上。常指由於環境因素等(如孕婦妊娠初期感染風疹等)而引起的疾病,它對以後的同代個體的發病一般無影響,預期風險近似整個群體的風險。
b.輕度風險:發病機率1∶10~1∶20。常指多基因遺傳病的復發風險,要根據該病的遺傳度、閾值等進行綜合分析計算。
c.高度風險:發病機率為1∶1~1∶10。所有單基因遺傳病(常染色體顯性遺傳病,隱性遺傳病,X伴性遺傳病)以及雙親之一具有平衡易位染色體的情況均屬此類
B.遺傳病的復發風險率估算:隨不同遺傳方式的遺傳病和已知信息的不同而定。在單基因的復發風險估算中。分基因型已推定者和基因型未能推定者兩種情況進行估算。
a.基因型已推定者:常染色體顯性遺傳病因患者多為雜合體若外顯率為100%,雙親一方為患者,則子女得病機率為50%,若已生育1個或幾個患者,復發風險仍為50%雙親均為患者時,子女的復發風險為75%,雙親皆非患者的子女一般不發病。突變個體的子女復發危險率為50%其同胞發病率等於群體自然突變率。若外顯不全則子女得病機率為K/2(K為外顯率,即實際觀到的患病數與預期值的百分比)。
常染色體隱性遺傳病:子代發病風險與雙親的情況關係密切。若是近親婚配子女患病風險提高。
X伴性顯性遺傳病:男性患者與正常女性婚配其子女中男性正常,女性發病。男性正常與女性患者婚配的子女各有50%發病。
X伴性隱性遺傳病:男性患者的兄弟有50%可能發病,其姐妹不發病,但有50%是攜帶者,同胞中總發病率為25%;其子女一般都不發病,但女兒為攜帶者,女性患者的子代中男孩100%發病,女兒都是攜帶者。男性患者與女性攜帶者婚配,其子女中各有50%發病。女性攜帶者與正常男性婚配,則子女中男孩有一半發病,女孩有一半為攜帶者。
Y伴性遺傳病:一般表現為父傳子,子傳孫僅限男性發病。
b.基因型未能推定者:若雙親之一或雙方的基因型未知,則估計發病子女或以後出生子女復發風險複雜得多。因為遲發外顯性遺傳病,雜合體要到一定年齡才發病。一個健康子女可以是完全正常的,也可能是一個尚未發病的雜合體要估計復發風險就必須推定其為雜合體的機率。在隱性遺傳病家族中,表型正常的雙親,生下正常的小孩,也不能斷定他們不是遺傳攜帶者,因為,即使雙親都是雜合子,生育正常小孩的機率也有3/4。當然,他們生下正常小孩越多,是雜合子的機會越小。在這種情況下估計復發風險,可根據上代和下代的表現型、實驗事檢查的結果,用逆機率定律(Bayes定律)來推算。
多基因遺傳病的致病基因有多對,呈共顯性;每個基因作用微小但具有累加效應,而且除了遺傳基礎外,環境因素在多基因遺傳病的發病中起較大的作用。不同的多基因病的遺傳度不同,發病閾值也有差異推算復發風險率時,比較複雜。一般來說遺傳度比較高(70%~80%)的多基因病若群體發病率為0.1%~1%,則患者一級親屬的發病率近似於群體發病率的平方根除此以外。還應考慮幾個問題現舉例加以說明。
唇裂在我國的群體發病率為0.17%,遺傳度為76%,患者一級親屬發病率為4%,接近於0.17%的平方根;當1對夫妻生了2個唇裂患兒後,復發風險率就相應增加,發病率由4%增至10%;如果患者病情嚴重,其發病風險率比病情輕的將增高,一側唇裂患者的一級親屬發病率為2.6%而兩側唇裂並齶裂者,復發風險率可高達5.6%,當發病率有性別差異時,發病率低的某性別患者的一級親屬復發風險率比發病率高的性別患者的一級親屬的復發風險率要高;先天畸形在新生兒中占1%~2%,當已出生1個這類畸形兒時再次妊娠發生這類畸形的風險率即隨已有患者數增多而增高。
多數染色體病患兒的親代核型都正常由於生殖細胞發生過程出現染色體畸形而導致後代發病。這類患者的同胞再患該病的風險率與一般人群相同;高齡產婦或有明顯誘變因素接觸史的雙親復發風險率可顯著增高;染色體數目異常(如13、18、21等染色體的三體性綜合徵),如果雙親之一的核型為嵌合型,則再生患兒的風險率可用下式估計P=[X/(2-X)]÷K(P為風險率,X為三體細胞的百分數K為係數,通常為2),染色體結構畸變所致疾病的復發風險率的計算,需根據不同的畸變類型,分析可能出現的分離、交換形式最後根據分離定律和交換定律分析配子的情況,才能得到估算。
③提出對策和解答問題:遺傳諮詢工作在詳實地了解病史及家系情況,分析了遺傳方式和估計了復發風險率後要對患者及其家屬提出的問題加以解答,對遺傳病的治療預防等問題提出對策並給予婚姻、生育等方面的指導。以切實起到預防遺傳病發生,造福人類的作用。
(3)產前診斷:產前診斷(prenatal diagnosis)又稱為宮內診斷(intrauterine diagnosis)或出生前診斷(antenatal diagnosis)是通過對孕期胎兒性別及健康狀況的檢測,以便及時採取必要的措施防止遺傳病或先天畸形患兒的出生。產前診斷是生化遺傳學細胞遺傳學、分子遺傳學和臨床實踐相結合的產物,當今高解析度顯帶技術、基因工程技術以及絨毛吸取和培養技術的發展,使產前診斷套用面更廣,檢查結果更準確。
①產前診斷的對象:
產前診斷常遇到的遺傳病有下列3類:
第1類:染色體異常,約占出生總數的0.5%由於容易明確診斷。故可占產前診斷病例的1/4~1/2。
第2類:單基因病,一般占出生總數的3.5%,占產前診斷病人的10%
第3類:多基因病包括無腦兒、脊柱裂、腦積水、某些唇裂和齶裂及某些先天性心臟病等。主要是神經管畸形,占產前診斷病例的40%~50%。
產前診斷適應證的掌握,不同國家、不同醫院有些區別,一般衛生保健條件好的地區和醫院掌握較寬通常普遍接受的適應證包括以下幾條:
A.35歲以上的高齡孕婦
B.夫婦一方有染色體數目或結構異常的孕婦。
C.已生過21三體綜合徵或其他染色體異常患兒及有相應家族史的孕婦
D.具有脆性X染色體家系的孕婦。
E.夫婦一方是染色體平衡易位或其他染色體畸變的攜帶者或嵌合體的孕婦。
F.夫婦一方是某種遺傳病患者,或曾生育過某種遺傳病患兒的孕婦。
G.夫婦一方有神經管畸形,或生育過開放性神經管畸形兒(無腦兒、脊柱裂)的孕婦。
H.有原因不明的自然流產,死產、新生兒死亡等病史的孕婦。
I.在妊娠早期曾受較大劑量的輻射或受病毒感染、長期服藥的孕婦
J.羊水過多的孕婦。
K.夫婦一方有明顯的環境致癌、致畸致突變因素接觸史的孕婦。
②產前診斷的方法:產前診斷按檢查對象不同,可以分為母體篩檢和胎兒檢查。其中母體篩檢可以有母體血血清甲胎蛋白篩檢、母體循環血中胎兒細胞檢查等。而通常所指的產前診斷主要是對胎兒的檢查診斷。它可以在不同水平上進行,選用不同的方法
A.形態學水平(表型水平):主要檢查胎兒有無先天畸形,常用的手段有:
a.X線檢查:妊娠16周后,胎兒四肢長骨、短骨、肋骨等都已經骨化可以通過X診斷畸形。在必要時,可用水溶性或油溶性造影劑注入宮腔,進行羊膜腔造影。
b.超聲診斷:超聲診斷是一種簡便而且損傷極小的產前診斷方法。常用的超聲診斷儀有A型超聲診斷儀,B型超聲診斷儀,超聲多譜勒診斷儀和M型超聲診斷儀。B型超聲診斷儀(B超)具有光點反差大,圖像清晰分辨力高等優點。多探頭電子自動快速掃描提高了掃描速度,可以直接觀察胎心。胎動等動態並可攝像記錄分析。B超常用於檢出:多胎妊娠;胎盤定位;性別鑑定;神經管畸形;內臟畸形;胎兒有核紅細胞增多征;胚胎髮育異常;宮內生長遲緩。
c.胎鏡:胎鏡(fetoscope)是一種帶有羊膜穿刺的雙套管的光導纖維內窺鏡。插入羊膜腔後可直接觀察胎兒畸形,也可採集胎兒活體組織和胎血、絨毛等材料,還能開展某些宮內治療,為遺傳病的預治提供了新途徑,但此操作可引起流產羊膜炎、母體免疫反應等並發征,故其套用受到一定的限制。
B.染色體水平:對宮內胎兒作染色體檢查,能早期診斷和預防常見的染色體病脆性X染色體綜合徵、染色體斷裂綜合徵以及與染色體異常相關的惡性腫瘤,也可以預測胎兒的性別有利於性連鎖遺傳病的預防。常用的材料是羊水中脫落細胞與絨毛細胞,一般經組織培養後,製備染色體片進行核型分析及高分辨顯帶等檢查,還可直接作X、Y小體檢查。絨毛細胞也可作短時培養後立即製片觀察。現有資料表明,絨毛細胞染色體分析結果和羊水細胞染色體分析結果不盡一致歐洲21箇中心1401例檢查的總結指出:a.絨毛的異常染色體率比中孕期羊水細胞高;b.出現的常染色體變異總數比中孕期羊水細胞高三倍,並發現一些不能存活的三體(如14、15、16三體);c.性染色體異常率也3倍於羊水細胞,其中45X比羊水細胞高10倍;d.夫婦之一有染色體平衡易位者其絨毛染色體有不平衡易位者也高於羊水細胞;e.絨毛細胞發現染色體異常的復發風險率為4.16%,羊水細胞為1.5%。早孕期絨毛細胞與羊水細胞診斷不完全一致至少能提示,在胚胎髮育過程中.自然界的選擇作用不斷淘汰異常細胞,而其他的意義尚不清楚。
C.酶學水平:通過對羊水及其中的細胞、絨毛細胞或母體血尿的酶學檢查可以檢出許多先天性酶異常疾病。
D.代謝產物水平:檢出特異性代謝產物,可預先診斷某些遺傳性代謝病,如黏多糖病等。
E.基因水平:利用羊水細胞、絨毛細胞或胎兒活檢組織甚至母體外周血中的胎兒細胞作材料,運用極度敏感和特異性很強的基因診斷技術,檢出遺傳病患兒
近幾年,隨著體外受精、胚泡體外培養、顯微操作取單細胞、人工胚胎移植等技術的發展出現了一種植入前診斷技術(又稱著床前診斷)。它套用現代分子生物學技術中PCR、原位雜交等靈敏度、特異度都很高的檢測手段,分析體外受精的胚胎或經子宮灌洗所得的胚泡中的單細胞或幾個細胞的基因組成,以明確是否為致病基因攜帶者,並將健康的胚胎移植入母體繼續發育這是產前診斷技術的又一發展但目前尚不成熟,未能推廣套用。
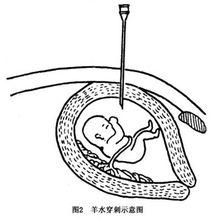
③羊膜穿刺術:受精卵發育第7天形成羊膜腔,生成羊水,羊水與胎兒直接接觸。是胎兒發育所需營養的主要供應途徑之一,也是胎尿的排出地,它的成份很能反應胎兒的生長代謝情況所以,羊水及羊水中脫落的胎兒細胞是產前診斷的主要材料,羊膜腔穿刺術的成功套用,是標本採集的關鍵。以下介紹羊膜腔穿刺術的技術要點
A.羊膜腔穿刺的適應證與禁忌證:凡是臨床及其他信息提示需要進行產前診斷的孕婦均為羊膜腔穿刺的適應證一般認為的禁忌證有:a.妊娠不足12周(子宮太小)或超過24周(細胞培養不易成功);b.適應證不明確;c.先兆流產或稽留流產的孕婦;d.盆腔或宮內有感染者;e.單純因社會習俗要求預測胎兒性別者
B.羊膜腔穿刺的時間:最好在妊娠16~20周。理由是:a.此時羊水量大(超過170ml),增長快抽出20ml羊水,不會因官腔驟小而引起流產;b.胎兒與羊水比例較合適胎兒小羊水多,周圍有較寬的羊水帶,穿刺不易傷及胎兒;c.羊水細胞中有活力的細胞比例此時最高,易培養成功;d.以上皮細胞和成纖維細胞為主的羊水細胞適合酶和生化分析。
C.穿刺的方法:穿刺前需認真作好以下準備工作:
a.核對適應證、妊娠周數、子宮大小有無併發症。
b.作外周血白細胞、血紅蛋白及血型檢查。
c.檢查穿刺部位的皮膚有無皮炎、感染等不利於穿刺的情況。
d.選擇好合適的穿刺部位,可以用B超來幫助定位胎盤及確定是否為單胎穿刺時也可在B超引導下進行
e.穿刺前孕婦必須排空尿液最合適的穿刺部位在恥骨上3橫指、腹中線旁最好宮底在臍恥之間,或臍下2橫指處,使進針部位剛好在子宮的中心或稍偏下進針前應仔細觸診。穿刺採用21號長腰穿針頭(帶針芯)。一般步驟:消毒穿刺部位及周圍皮膚,鋪洞巾,局麻,垂直快速進針,刺入皮膚後緩慢進針至7~8cm深(進入宮腔時可有落空感),抽出淺黃色透明液體,即為羊水(圖2)。先抽1~2ml用作生化檢查,沉澱細胞可用作性染色質檢查,另抽15ml放入無菌試管,用於作細胞培養。
D.羊膜腔穿刺中的常見問題:
a.穿刺失敗:一般的失敗率僅為0.5%~1%。可能的原因有:子宮太小,或穿刺部位太低誤穿了膀胱內的尿液;腹壁太厚進針不夠深;穿刺到胎盤附著部位,抽出血液後未敢繼續進針再抽。
b.羊水帶血:如開始即抽出血性羊水,提示針尖尚有部分在宮壁,宜將針深刺,抽出羊水後換乾淨的針筒再抽,即可順利抽出透明羊水。如果羊水抽得很通暢,而始終帶血則可能針尖刺中胎體或胎盤造成出血而致,胎盤前置的孕婦發生的可能性更大
c.對孕婦及胎兒的損傷:一般極少發生偶見有刺傷孕婦腹壁下動脈,形成大血腫而休克;穿刺在胎盤上,形成胎盤後血腫而致流產;穿刺傷及胎兒皮膚,出生後胎兒身上有點狀凹痕;刺傷胎兒致使一下肢壞死等報導。
d.官腔內感染:因操作時失誤,將細菌帶入官腔,可引起宮內感染和胎兒死亡故操作時要極端小心,嚴格無菌觀念。
e.流產:一般發生率極低。可能由於穿刺針眼羊水外流出血導致流產
f.Rh血型問題:Rh陰性血型的孕婦疑有胎兒Rh血型不合時,可在穿刺後給孕婦注射抗D球蛋白,如果胎盤在後壁附著,則可不必。
g.羊膜腔穿刺術的套用:表2列出了羊膜腔穿刺術在產前診斷中的套用範圍在此僅對羊水細胞培養的技術細節和羊水的生化檢查作進一步的說明。
h.羊水細胞培養的注意事項:羊水細胞培養是為了獲得更多的胎兒細胞,以滿足其他檢查的需要。羊水細胞大部分為羊膜上皮及胎兒脫落上皮細胞要培養成功,應注意:有活力的細胞要有一定的比例;培養基的品種和小牛血清要經選擇,HamF10,HamF12培養液成功率較高,小牛血清或牛胚胎提取液含生長素,對促進有活力的細胞生長很重要;促進羊水細胞及早貼壁,一般貼壁時間為5~7天,開始貼壁時多為上皮細胞,換液後成纖維細胞有大片生長;注意影響培養細胞成活的因素。培養細胞生長太快或太慢都要考慮母體細胞的污染;離體細胞培養時,會發生染色體的變異。需加以鑑別;其他影響因素:真菌、支原體污染抗生素的使用羊水中血細胞的污染及培養的其他條件的影響。
i.羊水的生化檢查:羊水的生化成份的變化可以直接反映胎兒的生長發育情況,對其作細緻的生化分析,可以提供很多遺傳病的信息,此類研究現在已越來越多。表3列出了幾種常見的羊水生化指標與其相應的遺傳病。
④產前診斷的意義:產前診斷能在胎兒出生前預先明確是否患有某種遺傳性疾病或先天畸形,通過精確的染色體分析和基因診斷還可明確是否為某種遺傳變異的攜帶者,為進行臨床疾病防治及遺傳病的各級預防提供最直接的依據。根據臨床資料、輔助檢查資料群體調查資料及產前診斷結果可以進行綜合分析,及時採取必要措施如對患病胎兒的選擇性終止妊娠,對可作早期治療的遺傳病(如苯丙酮尿症)進行治療,對有些簡單的先天畸形還可進行宮內手術治療。產前診斷已成為現代優生學的重要基礎方法,它在幫助限制整個人群致病基因的擴散,降低遺傳病發生率,監控出生人群的遺傳素質等工作中起著越來越大的作用。隨著醫療保健條件的改善,產前診斷的適應證不斷擴大,也為醫學遺傳學的研究提供大量的第一手資料。
2.艾-盪綜合徵的二、三級預防 從遺傳病預防學角度看遺傳病的治療屬於二級和三級預防的範疇。遺傳病的治療的關鍵是:儘早發現儘快治療。治療時機的掌握主要有以下幾種:①出生前確診(產前診斷)後,可進行產前治療(宮內治療)或產後立即治療。宮內治療方法有孕婦給藥療法和直接治療胎兒兩類。只要所使用的藥物能通過胎盤,孕婦給藥法就方便、安全,易被接受。如孕婦服用生物素、維生素B12、腎上腺皮質激素、洋地黃等,可分別治療胎兒的生物素依賴性羧化酶缺乏症、維生素B12依賴性代謝性酸中毒、先天性腎上腺皮質增生症和先天性室上性心動過速。對不能通過胎盤的藥物,可直接注入羊膜腔,讓胎兒在吞咽羊水過程中將藥物一併吞食。如甲狀腺素直接注入羊水可治療遺傳性甲狀腺腫。胎兒外科手術治療,現也有成功的報導;②典型症狀出現前予以確診(症狀前診斷)確診後給予儘早治療。例如苯酮尿症患兒可在出生後吃奶72h後用Guthrie血斑濾紙細菌抑制法進行早期確診,給予低苯丙氨酸飲食治療可以防止患兒智力損傷;③各種症狀都出現後才被確診。此時器官組織的損害都已出現,治療方法便不多,而且療效欠佳。可採取外科手術(病損器官切除、修補替換等)和內科對症療法來改善症狀。
遺傳病治療中總的原則是禁其所忌,去其所余,補其所缺,調節代謝平衡,防止症狀的出現。
(1)糾正代謝紊亂:這是目前治療遺傳性代謝病的最主要方法,隨著對遺傳性代謝病發病機制和中間過程的認識不斷深化,此法的適用範圍也日益擴大。
①飲食控制(禁其所忌):當代謝異常造成機體某些必須物質缺乏時,通過飲食加以補充;而當代謝物質發生貯積時則限制此代謝物或其前身物質的攝入來維持平衡。苯酮尿症患者低苯丙氨酸飲食就是很好的範例另外,還可通過限制對特定物質的吸收來減少攝入,如苯酮尿症患者服用苯丙氨酸氨基水解酶膠囊,可以將食物中的苯丙氨酸轉化為轉苯丙烯酸,而被消除。
②減少底物(去其所余):因代謝產生有害物質而引起疾病時,可以通過降低有害底物和減少其前身物質及代謝衍生物的濃度,去除或減少其毒性作用來控制或改善疾病的症狀。主要方法有:A.螯合或促進排泄;B.血漿置換法和親和結合法;C.改變代謝途徑;D.外科旁路手術;E.代謝抑制。
③產物替代(補其所缺):當重要的酶促反應產物不足而致病時,可直接補充相應的必需的終產物如給垂體性侏儒患者以生長激素,給血友病患者以抗血友病蛋白(凝血因子),給遺傳性免疫缺陷病人以相應的免疫球蛋白。
(2)糾正酶活性異常:
①輔酶的補充:有些遺傳病,酶活性異常可能累及:
A.一種特異性輔酶或維生素的結合部位。
B.有活性的輔酶轉運或生物合成過程導致異常許多輔酶是全酶正常活性所必需的。所以補充輔酶成分也是誘導酶活性增加的一種有效方法,它可以使全酶在細胞內降解速度減慢提高酶的半壽期,還可降低酶促反應的米氏常數(Km),目前已用此方法治療25種以上的遺傳病。如用鈷胺素(B12)治療多種貧血和甲基丙二酸尿症等。
②酶誘導或反饋抑制:對酶缺陷水平的另一種療法是用藥物來提高殘餘酶活性以改善代謝水平。例如苯巴比妥和有關藥物能明顯刺激滑面內質網的生成,並能加速內質網中特異性酶合成,包括肝UDP葡萄糖醛酸轉移酶,為用苯巴比妥治療Gibert綜合徵和Crigler Najjar綜合徵提供了理論基礎。
反饋抑制作用是許多代謝調節中的重要形式,針對因某種酶缺陷引起的底物或其前體堆積,可以通過其他旁路代謝的反饋抑制作用來提高酶活性,減少堆積的底物,反饋抑制已作為治療急性卟啉症的一種方法。
③同種移植:通過向遺傳病個體植入同種含正常基因的細胞,組織或器官以期在受體內產生相應的有活性的酶及其他基因產物,達到治療目的。移植物在受體內可能通過兩種機制發揮作用:
A.產生活性酶,在原位代謝除去原來的貯積底物。
B.釋放活性酶、輔酶或免疫活性因子入血,分布到全身其他組織中發揮作用。至今已進行過此類同種移植的組織器官有:腎肝、腎上腺骨髓、胸腺、脾胰等,有的已取得明顯療效。
④酶替代療法:直接給酶缺陷患者提供相應的正常的酶。隨著酶學技術和細胞工程、基因工程技術的發展,已經可以提供足量的、高純度的酶製劑。這種酶製劑必須具有半衰期長、抗原性低、導向性好等特性。為此常採用的方法是:
A.採用微囊、脂質體、紅細胞影泡等載體來包裝酶製劑,以減小免疫原性,延長半衰期
B.套用受體介導分子識別法來提高導向性。
C.對一些溶酶體貯積病因其沉積物可以彌散入血,並保持動態平衡則可用“平衡一去除”法來治療。
(3)基因治療:基因治療是指運用基因轉移技術直接將遺傳物質導入生殖細胞或體細胞以起到對遺傳病及其他疾病的治療作用的新型治療方法。對遺傳病進行基因治療可望從根本上糾正遺傳病的表型異常。
①基因治療的基本策略:近10餘年來,基因治療研究蒸蒸日上提出了許多新思路、新構想,目前主要的策略有:
A.基因的原位修正(correction)和原位替代(replacement):這一策略的目的就是要將突變的基因在原位修復,而不影響其周圍其他基因的結構和功能。其中原位修正針對基因的點突變或小範圍變異,擬通過特定方法對其定點修復。而原位替代,就想把有較大範圍變異的基因去除而換之以正常的基因。這一策略是最理想、最直接的對遺傳變異進行根治的方法,目前研究很多的哺乳動物細胞內定點整合(同源重組)給這種策略提供了理論和實驗依據,但至今未能真正用於人體試驗。
B.基因增強(gene augmentation或gene complementation):在不改變缺陷基因本身的前提下,將外源有功能的基因轉移到疾病細胞或個體基因組內,使其表達以補償有病基因失去的功能。此策略是目前研究最多,也是最成熟的方法。
C.將反義基因或其他對抗異常基因表達產物的基因導入細胞內起到抑制作用或稱基因抑制療法(gene inhibition therapy)或細胞內免疫(intercellular immunity)。
②基因治療的技術要點在基因治療的諸多策略中研究最多、最成熟並套用於臨床試驗的是基因增強的策略整個研究過程通常包括臨床前研究和臨床研究見表4。
A.疾病的選擇:目前基因治療首選的是單基因缺陷性疾病。選擇的基本條件常包括:
a.遺傳基礎比較明確,目的基因能在體外克隆
b.基因表達不需精細調節而且經常開放,產物生理水平不高者更佳。
c.具有一定發病率,危害較大,尚缺乏其他有效治療措施者。
中國是開展基因治療研究較早的國家之一,復旦大學薛京倫等就是根據這些條件選擇血友病作為研究對象,已取得了很好的結果,達到了世界先進水平。當然這些條件是限於現有的研究水平才提出的
B.靶細胞的選擇:基因治療的靶細胞可分為兩大類:生殖細胞和體細胞。由此引出了生殖細胞基因治療和體細胞基因治療的分類。如果能對生殖細胞或早期胚胎細胞進行基因修復或替換使基因缺陷得到校正,使遺傳病不但能在當代得到治療,還能將新基因傳給下一代,也為人群減少一個有害基因,是理想的遺傳病根治手段。但是由於現代生物技術、理論的限制,以及生殖細胞基因操作涉及人類社會的倫理、道德和法律等多種因素,在相當長的一段時間內只能進行動物試驗1985年美國政府就已規定,把基因治療的人體試驗限制在體細胞。已經被用於作為靶細胞的有:造血幹細胞、肝細胞成纖維細胞內皮細胞淋巴細胞等。
C.基因轉移的載體和轉移方法:構建合適的轉移並表達的載體和選擇高效的基因轉移方法是基因治療的關鍵,常用的載體有:逆病毒載體、質粒載體和腺病毒載體,腺相關病毒載體,另外還有脂質體載體。常用的基因轉移方法有4大類型。
a.化學法:主要是磷酸鈣沉澱法。
b.物理法:常用電導和顯微注射法。
c.膜融合法:以脂質體包裹法較好
d.病毒法:主要指逆轉錄病毒和腺病毒介導的基因轉移。
③基因治療的前景:基因治療概念的提出已有幾十年的歷史只到了近十年,隨著現代分子生物學技術(特別是DNA重組技術)的發展,這一概念才得到有力的理論基礎和技術方法的支持,並得以付諸實施1990年2名腺苷脫氨酶(ADA)缺陷引起嚴重免疫缺陷的患者接受基因治療獲得成功這標誌著基因治療的研究進入了一個新的階段。從此世界各國的生物醫學家在各國政府部門及社會各種力量的大力支持下全面展開了基因治療的研究。由原來針對單一的遺傳病發展到腫瘤、傳染病等多種疾病,提出了基因調控療法、基因抑制療法等新概念、新途徑。到1994年上半年,已有100多個臨床試驗方案獲準實施,有的已取得很好的效果。當然基因治療發展的歷史還不長,要廣泛套用於臨床還需大量的研究探索,尤其是以下幾方面的問題:
A.對更多遺傳病的分子基礎及基因表達調控機制更深入的了解,這是基因治療的基礎
B.構建更有效和安全地表達並轉移的載體。
C.更簡便有效的基因轉移方法的建立。
D.定點整合、原位修復系統等技術的完善。
E.更多更接近實際的動物模型(尤其是轉基因動物模型)的建立,這是基因治療臨床前試驗的必由之路。
F.體細胞基因治療生殖細胞基因治療等的倫理學及相關的科技管理立法等方面的探討。
G.還需充分考慮基因治療可能存在的危害性,如插入突變導致的嚴重後果、缺陷病毒載體經重組後恢復感染性的危害及外源基因轉入體內的其他潛在危害等。總之,我們認為基因治療作為一種惟一從基因缺陷本身入手,可望徹底治療遺傳病新型治療途徑,有非常吸引人的前途,但仍需從基礎理論、技術方法及倫理道德等多方面進行深入廣泛的研究探索,才能適應現代醫學模式被人們所接受,真正成為人類防病治病的有效手段
罕見病詞條庫

盤點常見的遺傳病
| 隨著分子生物學的發展,科學家們成功的把基因和疾病聯繫在一起。人們可以預先通過基因篩查來預測可能罹患的遺傳病。比如,GOOGLE公司創始人之一謝爾蓋?布林,就在其妻子的23andme生物技術公司通過測序發現自己可能在未來患帕金森氏綜合症,他積極通過各種方式來預防疾病的發生並投入巨資進行相關的研究。但如同一把雙刃劍,基因篩查也使一些攜帶突變基因的人在社會各個方面受到歧視,讓我們來關注一下遺傳疾病吧! |
| 黑棘皮症| 短趾症| 家族性高膽固醇血症| 白化病| 苯丙酮尿症| 紅綠色盲| 抗維生素D佝僂病| 先天性髖關節脫位| 脊柱裂| 先天性聾啞| 蠶豆病 | 強直性肌營養不良| 亨廷頓氏病| 老年性痴呆症| 精神分裂症| 脆性X綜合徵 | 原發性高血壓| WILSON 氏病| 成人多囊腎病| 馬凡綜合徵 | 侏儒綜合徵| 視網膜色素變性| 糖原貯積症| 神經鞘脂貯積症| 黏多糖貯積症 | 半乳糖血症| 白癜風| 性反轉綜合徵| WILMS 瘤| 視網膜母細胞瘤| 成骨不全病| 自毀容貌症| 家族性多發性結腸息肉病| 強直性脊柱炎| 假肥大性肌營養不良| 牛皮癬| 多囊腎| 脆骨病| 神經纖維瘤| 上瞼下垂| 類風濕性關節炎| 癲癇| 先天性心臟病| 高膽固醇血症| 唇裂| 齶裂| 魚鱗症| 多指| 著色性乾皮病| 腓肌萎縮症| 並指| 畸形足| 青光眼| 全身自化| 結腸息肉症| 先天聾啞| 原發性小睪症 |
