概述
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病檢查
1.測定血清α1-抗胰蛋白酶濃度(正常值2000~3000mg/L):比正常減少10%~15%對診斷可能有幫助但不能確診因在急性炎症時血清α1-抗胰蛋白酶濃度可能增加2.pi表型分析套用等電聚焦或酸性條件下瓊脂電泳鑑定α1-抗胰蛋白酶表型可建立診斷目前PCR技術已用於檢測α1-抗胰蛋白酶變異體此法不僅迅速敏感性高而且只需極少量的細胞物質該技術對確定診斷人群篩檢及出生前診斷等均有用
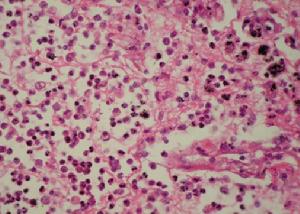
肝穿刺活組織檢查:顯示肝硬化PAS染色可見肝細胞內特徵性包涵體螢光染色顯示在肝細胞內蓄積有藍色顆粒即α1-抗胰蛋白酶抗體螢光帶
體徵
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病生理
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病Fagerhol等認為,控制α1-AT合成的所謂Pi基因是位於常染色體上的等位基因。採用薄層凝膠聚焦技術分析人類α1-AT電泳遷移率,發現其在人群中存在多態現象。現已鑑定出75種以上的α1-AT變異體,但大多數無臨床意義或很罕見,分別命名為B、C、D、E、F、G、L、M、N、P、S、V、W、X、Z等。各等位基因分別用PiM、PiS、PiZ等表示。純合子的基因型用PiMM、PiSS等表示,雜合子用PiMZ、PiSZ等表示。以上統稱為Pi基因系統。編碼α1-AT的基因定位於14號染色體長臂(14q24.3-32.1)。Pi基因系統的各種表現型,其血清蛋白酶抑制活性與α1-AT的濃度是不同的。PiM是具有正常功能的基因,絕大多數正常人是PiM的純合子(PiMM),其血清中α1-AT含量正常,功能也正常。具有PiZ基因的純合子(PiZZ)個體血清中α1-AT含量嚴重缺乏,僅為正常人的15%左右,這種人常發生阻塞性肺病和幼年型肝硬化。具有純合子PiSS血清中α1-AT含量中度缺乏,約為正常人的60%,這種人亦有患肺氣腫和肝硬化的傾向。雜合子PiMZ、PiSZ等個體也有發生肺氣腫和肝硬化的傾向。Jeppson等分析肽圖(peptidmapping)發現α1-AT缺乏症PiZZ變異型蛋白肽鏈中一個谷氨酸被賴氨酸所取代,一個谷氨酸被谷氨醯胺所取代,PiSS變異型系谷氨酸被纈氨酸所取代。α1-AT在肝細胞的粗面內質網產生,轉運到Golgi器供分泌之用。有一種假說,即與等位基因突變有關的蛋白錯誤摺疊(misfolding)構象,可能使α1-AT滯留在內質網而不能釋放到Golgi器。因為有此錯誤摺疊的變化,正常的隱蔽區可能暴露,從而與不同的配體受體接觸,而不能作為有效的分子釋放。異常的α1-AT滯留在內質網造成蓄積,排泌減少。其在細胞內的降解率取決於基因調控。α1-AT缺乏引起肝細胞損害的病理生理尚有爭議。目前認為肝損害使繼發於α1-AT在肝細胞粗面內質網的蓄積並有可能改變異常的α1-AT在肝細胞內的降解。α1-AT缺乏症患者的純合子和雜合子的肝細胞內質網內,可見過碘酸Schiff試驗(periodicacidschiff,PAS)陽性的耐澱粉酶顆粒,支持這一假說。α1-AT缺乏者有3個轉歸:一部分人可能終身健康;大部分人在青中年期患嚴重的肺氣腫;一部分人在嬰兒期就已患肝臟疾病。但是很少有同時患肺氣腫和肝硬化者。
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病尚不清楚為何一些人發生嚴重的肝病,而另一些人卻無症狀。多認為肝損害是由多方面因素引起的。如彈性蛋白酶能分解彈力纖維造成肺氣腫病變。但在正常情況下,彈性蛋白酶抑制因子可抑制此酶的活力,避免肺氣腫。研究發現PiZ較易發生慢性阻塞性肺疾病(COPD)。先天性α1-AT缺乏具遺傳易感性,需與後天外界因素結合才會產生致病作用。吸菸具有更大的危險性,如吸菸者肺巨噬細胞增多,細胞溶酶體多而大,菸草燃燒產生的NO2可刺激肺內巨噬細胞及中性粒細胞釋放彈性蛋白酶,而α1-AT缺乏者由於抑制蛋白酶的能力減弱,易發生肺組織損傷,從而引起慢性阻塞性肺疾病。α1-AT缺乏發生肝硬化者與肺部疾病無關。α1-AT缺乏是α1-AT缺乏性肝病的主要因素,還有其他因素參與,體內蛋白酶活性增高,是肝臟對其他一些致病因素和有毒物質的易感性增加,致使肝損害。Gam提出也有可能由於腸屏障破壞或有缺陷,腸內的毒素被吸收入肝,由肝Kupffer細胞攝取釋放出溶酶體酶,當人體缺乏α1-AT時該酶具有破壞性;或由於肝細胞內α1-AT滯留,腸毒素進入肝臟後,肝細胞內具有保護作用的蛋白溶解酶被過多的α1-AT抑制而使肝細胞受損;或因肝細胞內α1-AT過多而抑制了肝臟內源性蛋白酶的產生,以至不能對抗腸源性有毒物質,從而引起肝臟的損害。病理組織學改變因患者年齡而異。患病嬰兒的肝活標本檢查顯示膽管缺乏(bileductpaucity),肝細胞內膽汁淤積,伴有或不伴有細胞腫大變形,輕度炎性改變或脂肪變。肝細胞內可見一些特徵性的PAS陽性的耐澱粉酶樣小體(diastaseresistantglobule)。這種小體能被螢光素標記的α1-AT抗血清強烈染色,具有α1-AT抗原性。這種顆粒狀的包涵物位於肝細胞內質網上,隨著年齡的增長而增多。說明患者α1-AT的缺乏是由於合成的α1-AT不能釋放入血而蓄積於肝細胞內。以純合子PiZZ表現型患者為多。嬰兒期α1-AT缺乏的肝病患者,如無好轉可發生進行性肝損害。在門靜脈區纖維組織明顯增生,逐漸形成小葉間纖維化,可進一步呈小結節型或大結節型肝硬化。合子型α1-AT缺乏可引發原發性肝癌。
診斷
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病實驗室檢查:
1.測定血清α1-抗胰蛋白酶濃度(正常值2000~3000mg/L):比正常減少10%~15%,對診斷可能有幫助,但不能確診。因在急性炎症時,血清α1-抗胰蛋白酶濃度可能增加。
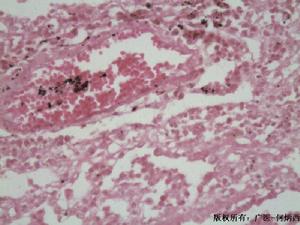
2.pi表型分析套用等電聚焦或酸性條件下瓊脂電泳鑑定α1-抗胰蛋白酶表型可建立診斷。目前,PCR技術已用於檢測α1-抗胰蛋白酶變異體,此法不僅迅速、敏感性高,而且只需極少量的細胞物質,該技術對確定診斷、人群篩檢及出生前診斷等均有用。肝穿刺活組織檢查:顯示肝硬化,PAS染色可見肝細胞內特徵性包涵體,螢光染色顯示在肝細胞內蓄積有藍色顆粒,即α1-抗胰蛋白酶抗體螢光帶。
治療
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病預防
 Α1抗胰蛋白酶缺乏性肝病
Α1抗胰蛋白酶缺乏性肝病2 肝損害嚴重者應行肝移植。由於肝是合成α1-抗胰蛋白酶的惟一場所,因此肝移植不僅能治癒肝病,且能糾正α1-抗胰蛋白酶缺乏。現認為肝移植是治療Pizz終末期肝硬化的有效方法,套用PiMM表型的供者肝臟做肝移植,可望提高其存活率及改善病情。
3 基因治療前景廣闊,但目前尚難奏效。糾正異常α1-抗胰蛋白酶的表達是預防肝損害的發生及控制其進展的關鍵。Zern採用特異ribozyrnes成功地抑制一肝腫瘤株異常α1-抗胰蛋白酶的表達,其抑制率達70%,這為預防α1-抗胰蛋白酶缺乏症肝病變的發生奠定了基礎。流行病學(查看內容)1977年Sever報導檢測20萬新生兒,觀察到120例為純合子(PiZZ)基因型個體,其中14例呈現長期膽汁淤積性黃疸。北歐白種人發病率相對較高,其純合子缺乏(homozygousdeficiency)發生率是1/1500,美國人是1/1800~1/2000,黑人、西班牙人和亞洲人較低。國內僅有少數個案病例報。
