
病因詳情
多發性神經纖維瘤又稱馮雷克林霍增氏病,為常染色體顯性遺傳疾病,系外胚層和中胚層組織發生障礙所致。
多發性神經纖維瘤特點是多系統、多器官受累而以中樞神經系統最為明顯。該瘤分兩型:NFI型的特點是皮膚的牛奶咖啡斑和出現神經纖維瘤樣的皮膚腫瘤。
多發性神經纖維瘤還常伴有多種畸形和其他一些疾病,譬如脊柱側凸、脛骨假關節、智力障礙、腦膜瘤、神經纖維瘤和眼的虹膜結節等,而且與神經纖維瘤病的惡性變傾向有關。
NFII型神經纖維瘤病是中樞型神經纖維瘤病,明顯的特點是雙側聽神經的神經鞘瘤、腦膜瘤和脊神經背根的神經鞘瘤;很少有皮膚改變。
臨床表現
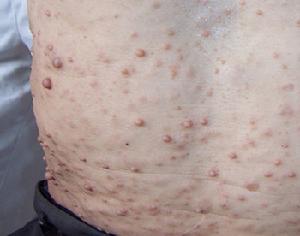
多發性神經纖維瘤見皮膚色素沉著,典型者呈牛奶咖啡斑,可為多發皮膚纖維瘤、雙側聽神經瘤。可伴有中樞神經系統的腦膜瘤、膠質瘤等。
皮膚轉移性癌最常見的臨床表現為皮膚或皮下結節,其色澤可與正常皮膚顏色相同,也可為紅色、淡紅或紫紅色,質地比較硬或韌。結節可與皮下組織粘連,但少有破潰。皮膚轉移癌通常出現於原發腫瘤診斷後,且多是惡性腫瘤已達到晚期的臨床表現,所以對出現在皮膚上的結節、腫塊一定要引起腫瘤病人的重視。尤其是那些僅有皮損表現,以轉移癌為首發症狀的惡性腫瘤患者。
影像學表現
多發性神經纖維瘤:NFIX線平片可見皮下多髮結節影,結節較軟,密度較淡。由於X線高空間解析度、CT的高密度解析度,在發現NFI的骨質病變上有著不可替代的優越性,如發現NFI型脊柱側彎、顱縫缺損、蝶骨翼發育不全等;NFⅡ型的內聽道口擴大或骨質破壞、繼發於髓內或神經根腫瘤的脊神經孔擴大或骨質改變等。MR的敏感性高,可進行多平面圖像顯示,無創傷,無放射性,可以發現比CT更多的顱內病變,顱內微小病變的檢出和病變大小的描述,則必須靠MR及MR增強檢查來實現,由於NF可導致中樞神經系統的多發病變,MR可見雙側聽神經瘤,多數以一側為重;多對顱神經、周圍神經的神經纖維瘤;合併腦膜瘤或膠質瘤。此外,NFⅡ型皮膚損害較少見,主要病變位於中樞神經系統,但當患者僅表現為多發腦(脊)膜瘤或多發神經纖維瘤時,即使沒有聽力異常,亦應常規進行頭MR及增強掃描,以進一步明確有無聽神經瘤。
皮膚轉移癌的診斷主要通過針吸細胞學檢查或手術活檢作出的病理診斷,並可有原發腫瘤病史。皮下轉移灶部位皮膚多呈淡紅色,為無痛性皮下類圓形結節或腫塊,高出皮膚,質地較硬,表面尚光滑,無破潰,活動性差,直徑1.5cm-7.0cm。其治療首先是針對原發病灶,對於皮膚局部腫瘤,可酌情採用手術切除、放射治療、冷凍或雷射療法。惡性腫瘤發生皮膚轉移臨床比較少見,發生率僅為0.7%-9%。開始是小紅點,如果在擴張的淋巴管或者血管內有轉移性的癌細胞存在。後逐步擴大。蔓延大片皮膚,連線成片,壓之退色,可成團塊狀丘疹,轉移的結節以多發為主,也可為孤立性的皮膚結節。大多數轉移的結節直徑小於3cm。而且60%多的皮膚轉移瘤伴有其他臟器的遠處轉移。一般認為惡性腫瘤一旦發生皮膚轉移,預後往往較差,治療效果不佳。對於皮膚轉移的治療,一般認為放療和化療均有一定的療效。但是總體的來說,不能明顯延長生存期。
診斷方法
多發性神經纖維瘤根據典型臨床表現、CT、MR表現等可明確診斷。
1、NFIX線平片可見皮下多髮結節影,結節較軟,密度較淡。
2、由於X線高空間解析度、CT的高密度解析度,在發現NFI的骨質病變上有著不可替代的優越性,如發現NFI型脊柱側彎、顱縫缺損、蝶骨翼發育不全等;NFⅡ型的內聽道口擴大或骨質破壞、繼發於髓內或神經根腫瘤的脊神經孔擴大或骨質改變等。
3、MR的敏感性高,可進行多平面圖像顯示,無創傷,無放射性,可以發現比CT更多的顱內病變,顱內微小病變的檢出和病變大小的描述,則必須靠MR及MR增強檢查來實現,由於NF可導致中樞神經系統的多發病變,MR可見雙側聽神經瘤,多數以一側為重;多對顱神經、周圍神經的神經纖維瘤;合併腦膜瘤或膠質瘤。
分析討論
多發性神經纖維瘤涉及神經、皮膚、骨骼系統,根據典型臨床表現、CT、MR表現可明確診斷。腫塊呈多發性、數目不定,少的幾個,多的可成百上千難以計數。小的如米粒,大的似拳頭。可鬆弛地懸掛於皮表,皺褶及鬆弛可致畸形明顯。神經纖維瘤沿神經乾的走向生長時呈念珠狀,或蚯蚓狀結節。此外神經纖維瘤皮膚可出現咖啡斑,大小不一,形如雀斑小點狀,或大片狀,分布與神經纖維瘤腫塊的分布無關。腫瘤數目不多的患者,皮膚色素咖啡斑狀沉著是多發性神經纖維瘤病的重要診斷;本病多發於軀幹,有時出現於四肢及面部,患者常合併許多疾病。
皮膚轉移性癌是各種惡性腫瘤轉移的部位之一,而原發性肺癌皮膚轉移較少見,文獻報導甚少,女性患者多來自乳腺癌(約69%),其次為大腸癌(9%),來自肺癌、卵巢癌和惡性黑色素瘤的占4%-5%。轉移性皮膚癌可發生於任何年齡組,但多數發生在40歲-70歲。Coslett指出胸壁、腋窩及腹部是皮膚轉移癌的好發部位,臨床上由於原發性肺癌起病隱匿,尤其周圍型肺癌常缺乏典型的呼吸道症狀,而常被延誤診斷,有時皮下轉移灶可能是肺癌的唯一特徵。因此,臨床上若遇不明原因的無痛性皮下結節,應及時行皮下結節病理切片,如為癌性病灶,應及時行胸部X線檢查或CT檢查,以確定原發灶是否為肺臟。

