
類型
據臨床表現骨骼、眼、心血管改變三主征分為兩型:三主征俱全者稱完全型;僅二項者稱不完全型。
流行病學
據估計在美國大約有60,000(占人口數0.02%)到200,000人患有此病。該病致病基因攜帶者有一半的幾率將其傳給下一代。大多數馬凡氏綜合症患者有家族史,但同時又有15~30%的患者是由於自身突變導致的——這種自發突變率大約是2萬分之一。
據心臟外科醫生稱,中國人患上此病的比率為每十萬人有十七人患病。由於沒有正式統計資料,實際患者比率可能更高。
研究歷史
 馬凡氏綜合症
馬凡氏綜合症1896年,法國小兒科醫生安東尼·馬凡(AntoineMarfan)對一個5歲女孩的診治過程中第一次發現並描述了該病的症狀。此後100年間人們逐漸認識到這是一種累及多系統的新的遺傳相關疾病。
1914年發現它可以合併有晶狀體異位。
1931年發現其因中胚層缺陷導致,是常染色體顯性遺傳病。
1943年發現可合併主動脈擴張和夾層動脈瘤,
1955年正式被歸類為結締組織遺傳病,
1975年發現合併二尖瓣脫垂,
1988年發現可有硬脊膜膨脹。
馬凡氏綜合症的診斷標準也幾經修訂。1979年,美國約翰霍普金斯醫學院的ReedE.P等人從心血管、眼、骨骼及家族史四個方面系統的闡述了馬凡氏綜合症的臨床表現和診斷依據。
流行病學
 馬凡氏綜合徵
馬凡氏綜合徵 馬凡氏綜合症發病無性別傾向,其突變率亦無地域傾向。據估計在美國大約有60,000(占人口數0.02%)到200,000人患有此病。該病致病基因攜帶者有一半的幾率將其傳給下一代。大多數馬凡氏綜合症患者有家族史,但同時又有15~30%的患者是由於自身突變導致的——這種自發突變率大約是2萬分之一。馬凡氏綜合症也是一個顯性負突變(dominantnegativemutation)和單倍不足(haploinsufficiency)的疾病例子。其發病與可變表現度(variableexpressivity)有關,但不完全外顯率(incompletepenetrance)還沒有被正式確定。
發病機理
本病原發缺陷不明,其發病與可變表現度(variableexpressivity)有關,但不完全外顯率(incompletepenetrance)還沒有被正式確定。
有人認為是彈性蛋白和膠原組織肽鏈之間的橫向聯合受損,即賴氨醯氧化酶缺陷。此外與酸性粘多糖沉積、唾液酸增多、透明質酸堆積、硫酸軟骨素形成不良或過度破壞有關。
人類第15號染色體上有一個叫做FBN1的基因,編碼微纖維蛋白1。它可以被看成是把身體細胞黏合在一起的膠水。這個基因發生突變後,“馬凡氏綜合症”便產生了。
臨床表現
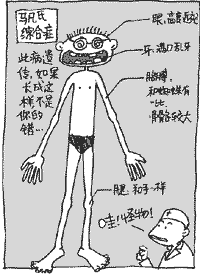 馬凡氏綜合症
馬凡氏綜合症2、眼:主要有晶體狀脫位或半脫位、高度近視、白內障、視網膜剝離、虹膜震顫等。男性多於女性。
3、心血管系統:約80%的患者伴有先天性心血管畸形。常見主動脈進行性擴張、主動脈瓣關閉不全,由於主動脈中層囊樣壞死而引起的主動脈竇瘤、夾層動脈瘤及破裂。二尖瓣脫垂、二尖瓣關閉不全亦屬本徵重要表現。可合併先天性房間隔缺損、室間隔缺損、法樂氏四聯征、動脈導管未閉、主動脈縮窄等。也可合併各種心律失常如傳導阻滯、預激綜合徵、房顫、房撲等。
診斷
在1996年修訂的馬凡氏綜合徵的診斷標準中,在骨骼、眼睛、心血管、肺、皮膚及體包膜、硬腦(脊)膜、家族遺傳史等方面分別列出了幾項主要標準和次要標準,無家族或遺傳史者,需有吻合兩個以上不同系統的主要標準並牽涉到三分之一以上的器官,才能確診其患有馬凡氏綜合徵。
診斷標準
 馬凡氏綜合症
馬凡氏綜合症•雞胸;
•漏斗胸,需外科矯治;
•上部量/下部量的比例減小,或上肢跨長/身高的比值大於1.05;
•腕征、指征陽性;
•脊柱側彎大於20度,或脊柱前移(側彎計);
•肘關節外展減小(<170度);
•中踝中部關節脫位形成平足;
•任何程度的髖臼前凸(髖關節內陷)(X片上確定)。
•馬凡氏綜合徵的患者,大多有高度近視。
骨骼作為一項主要標準,8項表現中必須至少有4項吻合方可考慮骨骼受累。
診斷
診斷此病的最簡單手段是超聲心動圖,有懷疑者均可行此檢查,進一步確診則需要通過MRI(磁共振顯像)
治療
目前(截止2013年)尚無特效治療。對先天性心血管病變宜早期手術修復,對心功能不全、心律失常者宜內科治療。一旦確診為合併有主動脈瘤或心臟瓣膜關閉不全,則應視情況考慮手術治療,因為藥物是不能去除此病的。
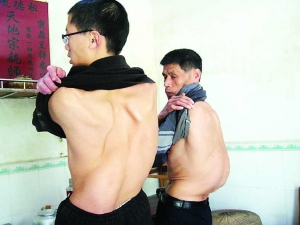
病例
二十世紀80年代與郎平齊名的美國女排主攻手海曼,1988年奧運會雙人滑冠軍格林科夫以及四川男排名將朱剛。NBL瀋陽東進隊中鋒武強,也因為此病於2009年在賽場上猝死。
2013年10月,中國男籃中鋒張卓君,在體檢時被查出患有馬凡氏綜合症而進行了手術,其職業生涯就此劃上句號。

