簡介
心臟變異型,是法布瑞氏症里較不容易早期診斷的變異型。
 症狀表現
症狀表現病因
 基因研究
基因研究法布瑞氏症的基因位於X染色體上,通常男性的病況較女性來得嚴重,在美國約有4萬分之一的男性罹患此疾病。臨床症狀通常在兒童與青少年期開始出現,典型的患者會出現肢體末端間歇性的疼痛、皮膚上呈現暗紅色斑點且多半分布於下腹部到大腿之間。到了成年之後,出現進行性的腎臟、心血管及腦血管病變,成為威脅生命的主因。
這種疼痛常有燒灼般的特性,當肢體疼痛來襲時,有些患者甚至痛到掉淚、呻吟、在床上翻滾,甚至用沖冷水來減緩疼痛。他們很可能因為疼痛而嚴重影響生活品質,甚至面臨休學、放棄工作的窘境。此病在早期很難被診斷出來,因此常被外人誤認為裝病而飽受委屈。除此,長期生活在不知名的疼痛恐懼中,也較容易有憂鬱傾向。
法布瑞氏症在過去僅能採取症狀治療,隨著基因工程技術的進步,使酵素替代療法成功的運用在法布瑞氏症的治療上。它不僅止於改善症狀,更象徵著可進一步延長壽命與提高生活品質。法布瑞氏症的患者若能及早診斷,並依照醫師指示接受治療,相信只要有心經營生活,生命必然充滿著活力與希望。
法布瑞氏症(Fabry Disease)是一種罕見的遺傳疾病,它與粘多醣症、高雪氏症等疾病同屬「溶小體儲積症」。溶小體如同人體細胞的垃圾場,在正常狀況下,溶小體含有多種酵素,可將蛋白質、粘多醣、醣脂質等物質消化分解成小分子,提供細胞回收再利用。法布瑞氏症則是由於缺少了一種溶小體酵素---A型阿法半乳糖甘酶(α-galatosidase A;簡稱α-Gal A),使得醣脂質,特別是globotriaosylceramide(簡稱GL-3)無法進行分解,於是堆積在全身許多細胞的溶小體內,包括:腎絲球與腎小管的上皮細胞;心肌細胞與瓣膜纖維細胞;背根神經節的神經元與自主神經系統;角膜上皮細胞;血管內皮、外皮、平滑肌細胞,最終引發各個器官的病變。
法布瑞氏症的基因位於X染色體上,通常男性的病況較女性來得嚴重,在美國約有4萬分之一的男性罹患此疾病。臨床症狀通常在兒童與青少年期開始出現,典型的患者會出現肢體末端間歇性的疼痛、皮膚上呈現暗紅色斑點且多半分布於下腹部到大腿之間。到了成年之後,出現進行性的腎臟、心血管及腦血管病變,成為威脅生命的主因。
這種疼痛常有燒灼般的特性,當肢體疼痛來襲時,有些患者甚至痛到掉淚、呻吟、在床上翻滾,甚至用沖冷水來減緩疼痛。他們很可能因為疼痛而嚴重影響生活品質,甚至面臨休學、放棄工作的窘境。此病在早期很難被診斷出來,因此常被外人誤認為裝病而飽受委屈。除此,長期生活在不知名的疼痛恐懼中,也較容易有憂鬱傾向。
法布瑞氏症在過去僅能採取症狀治療,隨著基因工程技術的進步,使酵素替代療法成功的運用在法布瑞氏症的治療上。它不僅止於改善症狀,更象徵著可進一步延長壽命與提高生活品質。法布瑞氏症的患者若能及早診斷,並依照醫師指示接受治療,相信只要有心經營生活,生命必然充滿著活力與希望。
臨床症狀
臨床症狀通常在兒童與青少年期開始出現,最顯著的症狀是間歇性的肢體末端疼痛與感覺異常。除此之外,下腹部與大腿間的皮膚可見暗紅色的疹子,出汗的能力減少,眼睛的角膜呈現輻射狀或螺鏇狀濁斑。隨著年齡增長,在成人時期出現腎臟、心血管、腦血管病變,最後進展為腎臟衰竭、心臟合併症、早發性中風。另外,部份的患者會呈現腸胃道不適的症狀。詳細的症狀說明分述如下:
男性患者症狀
疼痛:為最常見也是最早被發覺的症狀。疼痛的形式可區分為兩種:一種是肢端感覺異常,是指肢體末端經常性的灼熱、刺麻與疼痛感;另一種是疼痛危象,是指從肢體末端向外擴散,斷續發生的劇烈疼痛感,可持續數分鐘至數周之久。通常在運動、疲倦、情緒壓力、高溫環境、氣候變化下較易出現疼痛。疼痛感一般會隨年紀的增長而頻率減少,但亦有例外。
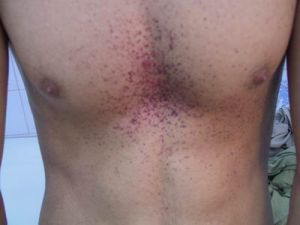
皮膚病變:是一種暗紅色的丘疹,大小從針狀到數公厘,多半分布於下腹、肚臍、臀部、陰囊、外生殖器、大腿部位,也可能出現於結膜、口腔與其他黏膜區域,稱為血管角質瘤。通常隨著年紀而有增加的趨勢,但也不一定出現。
排汗能力減少:因排汗減少而引起體溫過高、對天氣變化極端敏感。
眼睛病變:幾乎所有的男性患者與約70%以上的女性帶因者具有角膜濁斑的特徵,角膜上呈現奶油色的輻射狀或螺鏇狀變化。結膜與視網膜也可能受到影響,呈現程度不等的血管曲張。這些眼睛病變對大多數患者視力並不造成影響,但有些患者仍有視力受損的情形。另外,有某些患者出現晶狀體的變化,但較不常見。
腸胃道病變:容易出現腹脹感、進食後腹痛、嘔吐、腹瀉。
腎臟病變:腎臟功能將逐漸退化,於成人時期開始出現蛋白尿,腎絲球過濾率逐漸下降。若影響濃縮尿液的能力,則會出現多尿、口渴的現象。到了後期,進展為腎衰竭,是此病症最常見的致命合併症。
心臟病變:於成人時期開始出現病變,但嚴重度有個別差異性,包括:左心室肥大、二尖瓣閉鎖不全、心律不整。到了後期,常因冠狀動脈疾病而引發心絞痛、充血性心衰竭與心肌梗塞。另外,曾有國外報告表示,某些男性患者僅出現心臟病變,這類型的患者可能有輕微的蛋白尿,體內的α-半乳糖水解酶A仍具有少量活性,稱為心臟變異型。
腦血管病變:於成人時期開始出現病變,包括:腦血管梗塞、暫時性腦缺血、基底動脈梗塞、腦出血。常見的症狀有嚴重頭痛、單側麻木無力、視野缺損、昏眩或意識不清、平衡感喪失、說話含糊、表達困難、理解力降低等。
女性帶因者症狀
女性帶因者的症狀通常較男性患者來得輕微,其臨床表現差異極大,可能毫無症狀,也可能與男性患者同樣嚴重,這與女性X染色體的隨機不活化有關。約70%的女性帶因者具有渦狀角膜濁斑的特徵,部分伴隨有間歇性四肢疼痛或感覺異常、皮膚病變,少部分患者則出現心臟病變,僅極少數患者與男性患者同樣嚴重。
診斷原則
若有男性或女性出現不明原因的四肢疼痛、皮膚病變、排汗能力減少、特有的渦狀角膜濁斑、早期中風、左心室肥大或腎臟功能不全等症狀,應懷疑罹患法布瑞氏症。進一步可從家族史觀察家族成員是否有相似症狀或男性親戚間是否有早期的腎臟、中風及心臟疾病。相關臨床檢查包括:
初步診斷
腎功能檢查:尿液蛋白質與沉澱物、肌酸酐清除率、血液尿素氮與肌酐酸檢查。
心臟與腦血管功能檢查:胸部X光、心臟超音波、心電圖、核磁共振。
病理檢查:腎臟切片檢體。
確認診斷
酵素活性檢測
A型阿法半乳糖甘酶可藉血漿、血清、白血球或皮膚纖維母細胞培養檢測活性。男性患者的酵素活性小於1%,心臟變異型患者的酵素活性小於10%。女性帶因者的酵素活性個別差異大,與女性X染色體的隨機不活化有關,有些女性帶因者的酵素活性落在正常值範圍內。對男性患者而言,此法是極為有效與可靠的檢測工具,但對女性帶因者則無法完全辨別。
基因分析
法布瑞氏症的基因位於X染色體的長臂上,稱為GLA基因。至今,已有超過300個突變被確認出來,大部份的突變具有家族特異性,僅發生於單一家族。目前,基因分析已被運用於臨床診斷上,一旦有患者經基因定序分析確認突變,同樣的突變亦可用於找尋其他未發病的家族成員與女性帶因者。
產前診斷:女性帶因者可由產前檢查確認胎兒是否罹病,其檢測方法是在懷孕10-12周時進行絨毛膜採樣,或在懷孕15-18周進行羊膜穿刺,以獲取胎兒細胞。取得的胎兒細胞經培養後,通常會先作染色體分析以了解胎兒性別。若胎兒為男寶寶,則需作酵素活性檢測以確認是否罹病。若得知家族的突變,亦可直接進行突變分析。
治療原則
法布瑞氏症因涉及多種不同的系統,需要各科專家聯合診治。目前,此疾病的治療方式除了症狀治療以外,亦可採用酵素替代療法。在症狀治療方面,肢體疼痛可藉由一些藥物來緩解,若病情進展到腎衰竭的階段則需靠血液透析以維繫生命。在酵素替代療法方面,利用基因工程所合成的法布瑞氏症酵素製劑,經臨床試驗證實可有效減少醣脂質堆積於某些器官,有助於改善症狀與減緩長期合併症。至今,現有的兩種酵素製劑皆已經由衛生署核准為罕見疾病用藥。
酵素替代療法
Replagal (力甫蓋素濃縮注射液)
劑量:成人劑量為0.2 mg/kg,以靜脈輸注至少40分鐘,每兩星期接受一次靜脈輸注液。
相互作用:不可與Chloroquine、Aminodatone、Benoquin、Gentamicin一起使用。
注意事項:在一小時內之輸注過程中,約有10%患者產生輕微、急性之特異反應,一般症狀為寒顫及臉部潮紅。
副作用:發生頻率大於10%的症狀包括:血管方面-潮紅,胃腸道-噁心,皮膚部位-紅斑疹、座瘡,全身性症狀-灌注時可能發生胸痛、寒顫、發燒、疲勞、背痛、熱耐受力缺乏。
Fabrazyme (法布瑞凍晶注射劑)
劑量:16歲以上青少年及成人每兩星期接受一次靜脈輸注液,劑量為1 mg/kg。起初20個星期,藥品輸注速率為0.25mg/min。輸注時間不可少於2小時。
相互作用:不可與Chloroquine、Aminodatone、Benoquin、Gentamicin一起使用。
注意事項:對此藥過敏、中等至嚴重的高血壓(於輸注時可能惡化)、腎衰竭(對於血漿肌酸酐大於2.5mg/dL患者的研究不足)及發燒病患(會惡化)應避免使用。監控治療指標為定期測量血漿中GL-3濃度,正常濃度應低於1.2ng/ml。
副作用:發生頻率大於10%的症狀包括:全身性症狀-寒顫、對溫度變化敏感、發燒、四肢疼痛,呼吸系統-支氣管痙攣、喉嚨緊悶,胃腸道-噁心、嘔吐,中樞及周圍神經系統-頭痛、震顫,心血管-四肢水腫、高血壓,肌肉骨胳系統-肌痛。
遺傳諮詢
人體細胞內共有23對染色體,其中有一對可決定性別,稱為性染色體。性染色體可分為X染色體與Y染色體,男女的差別在於男性有一條X染色體與一條Y染色體(染色體核型以XY表示),女性則有兩條X染色體(染色體核型以XX表示)。
法布瑞氏症的基因位在X染色體上,屬性聯隱性遺傳,僅極少數為自發性新突變。男性因為只有一條X染色體,若有缺陷基因,便會罹病;女性有兩條X染色體,若一條X染色體有缺陷基因,只要另一條正常,通常病情較男性患者輕微,也就是所謂的帶因者。女性帶因者的臨床表現差異極大,這與女性X染色體的隨機不活化有關。X染色體不活化是指在女性,每個細胞內都含有兩條X染色體,其中會有一條X染色體不表現,此種現象是隨機的。若帶有缺陷基因的X染色體出現不活化的比例愈高,則病情愈輕。
以遺傳機率而言,若父親正常而母親是帶因者,則所生的兒子有50%的機率為患者,50%的機率正常;所生的女兒有50%的機率為正常,50%的機率為帶因者。若父親為患者而母親正常,則所生的兒子全部正常,所有的女兒為帶因者。所以,若有男性確認罹患此病,應配合醫師的建議,進一步了解家族成員的健康狀況,找出潛在的患者與帶因者,才能早期診斷早期治療。家族成員亦可透過遺傳諮詢取得相關遺傳知識,以作為選擇與決定的參考。

