基本簡介
貝賽特氏症(BD)作為一種系統性疾病,它牽涉的內臟器官包括胃腸道、肺動脈、肌骨骼、心血管和神經系統。由於貝賽特氏症可影響幾乎人體內的每個器官,其他情況如血管炎,纖維肌肉痛、偏頭痛或中樞神經系統的問題、視力問題、心跳過速和關節疼痛,腫脹通常也與貝賽特氏症有關。
貝賽特氏症在中緯度地區有較高的病發率,例如土耳其一帶,日本的發生率亦較其他國家來得高。台灣的全民健保把此疾病列為重大傷病之一。貝賽特氏症與基因HLA-B51有較高的相關性。
疾病來源
貝賽特氏症(BD)於1937年被土耳其的皮膚科醫生貝賽特(1889-1948)而命名,他亦是一名科學家,他於1924年是第一位從病人身上發現這種疾病,並在1936年在《皮膚期刊》發表這種疾病和性病的報告。
基本症狀
貝賽特氏症主要症狀為:反覆性口腔潰瘍(recurrentoralulcers)及口瘡三重複合症、反覆性生殖部位潰瘍(recurrentgenitalulcers)及葡萄膜炎(uveitis)。
皮膚與黏膜
幾乎所有患者都有某些形式的疼痛性口腔黏膜潰瘍、口瘡性潰瘍或無結疤口腔損傷。口腔損傷類似於那些在炎性腸道疾病中找到,並且可以是復發性。疼痛的生殖器潰瘍通常在肛門、外陰或陰囊周圍形成,並導致75%的患者結疤。此外,患者可能出現結節性紅斑,侵犯皮膚的膿皰性血管炎,病變類似同源染色體。
視覺系統
炎症眼疾會於發病早期形成,有個案導致失去20%的視力。視覺系統受損的形式可以是前葡萄膜炎、後葡萄膜炎或視網膜炎。前葡萄膜炎會呈現疼痛的眼睛、結膜充血、前房積膿、而且視力下降。同時,後葡萄膜炎會呈現出無痛性視力下降和飛蚊症視野。在貝賽特氏症,牽涉眼睛出現視網膜血管炎的形式是很罕見的,
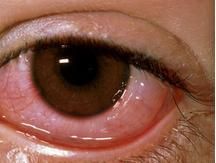 結膜充血
結膜充血當中視力可能在無痛的情況下出現下降及飛蚊症,以及視野缺損的情況。視神經萎縮已被確定為視覺障礙的最常見原因。貝賽特氏症會導致原發性或繼發性視神經受損,視神經乳頭水腫是靜脈竇血栓形成導致的結果及視網膜疾病萎縮引致,它已被定性為貝賽特氏症視神經萎縮的的次要原因。視神經病變的跡象與症狀包括無痛喪失視力會影響一隻或雙眼,使視力下降、降低了辨色能力、相對傳入性瞳孔缺陷、中央盲點、視神經盤腫脹、黃斑水腫及眼球後疼痛。當這些病症與皮膚黏膜潰瘍同時出現,醫生會提出貝賽特氏症引發的急性視神經病變的懷疑。視神經漸進性萎縮,可能導致視力和顏色視覺下降,那時顱內高血壓及視乳頭水腫可能會同時出現。
診斷標準
國際診斷標準
InternationalStudyGroupforBehcet’sDisease(ISGBD)。CriteriafordiagnosisofBehcet’sdisease.Lancet1990;335:1078-80.
1.反覆性口腔潰瘍:十二個月內至少復發三次。
以上加上下列任兩者
2.反覆性生殖器潰瘍:潰瘍或結痂(陰囊與陰唇為主)
3.眼部病變:前或後葡萄膜炎(uveitis),在玻璃體或細縫燈(slitlamp)下看到細胞,眼科醫師所見的網膜血管炎(retinalvasculitis)。
4.皮膚病變:結節性紅斑(erythemanodosum),假性毛囊炎(pseudofolliculitis),丘疹與膿皰樣疹(papulopustularlesions),或青春痘樣疹(acneiformnodules)。
5.陽性之pathergytest:
用20-22G針頭斜插到5mm深度,四十八小時後由醫師觀察(陽性>2mm)。
日本診斷標準
Behcet’sdiseaseresearchcommitteeofJapan:Behcet’sdisease:GuidetodiagnosisofBehcet’sdisease.JpnJOphthalmol18:291-292,1974
主要診斷要件
1.反覆性口腔潰瘍
2.皮膚病變
甲、結節性紅斑
乙、皮下靜脈血管炎
丙、毛囊炎,青春痘樣病變
丁、皮膚高敏感(Patergytest)
3.眼部病變
甲、虹彩炎
乙、網膜炎
丙、確定之虹彩炎網膜炎病史
4.生殖器潰瘍(男性主要在陰囊,女性主要在陰唇)
次要診斷要件
1.關節炎,無變形與粘連
2.胃腸道病變,尤其是回盲腸交接處(ileocecal)潰瘍
3.副睪炎
4.血管病變
5.中樞神經系統病變
用藥治療
眼局部按葡萄膜炎常規處理。糖皮質激素局部給藥,如0.5%地塞米松眼水頻頻點眼,睡前加用其眼膏。全身給藥以口服為宜,儘量避免靜脈給藥。原則上應大劑量短期使用,緩解後遞減漸停。與免疫抑制藥聯合套用,不僅可以增進療效,而且還能減少各自的劑量和副作用。
關於免疫抑制藥的選擇意見不一。在環磷醯胺(cyclophoshpamid)、硫唑嘌呤(azathioprine)、苯丁酸氮芥(瘤可寧,chlorambucil)等烷化劑免疫抑制藥中,以苯丁酸氮芥療效最佳。開始日劑量為0.1~0.15mg/(kg·d),持續5~6個月,或在炎症控制3~4個月後逐漸減量,最後減至2mg/d的維持量。整個療程在1年以上。用藥期間應定期檢查血象,若白細胞總數急劇減少,或眼底出現視盤水腫、視網膜出血等不良反應時應停用。另外,瘤可寧可引起不育症,用藥前應獲得患者及其家屬同意。腎功能不良者禁用。日本學者對環孢素的評價較高,重症病例之所以減少,與環孢素普遍套用有關。但也必須注意其毒副作用,肝、腎功能不良、降壓藥不能控制的高血壓、妊娠期患者,均屬禁忌,此外,環孢素A易於引起神經系統損害,因此對神經型Behcet病或有神經精神病史者,亦不宜套用。
免疫調節藥左鏇咪唑(levamisole)對口腔黏膜潰瘍、白細胞趨化因子抑制劑、秋水仙鹼對皮膚結節樣紅斑各具特點。對伴有這些眼以外病變的患者,可選擇使用。
貝賽特氏症多器官損害的病理基礎是血管周圍炎及血管內膜炎。實驗室檢查纖維蛋白溶解活性下降,凝血功能亢進,血液中免疫複合物增高,容易發生脈絡膜血管及視網膜靜脈血管阻塞等。因此,纖維溶解酶原激活劑、血小板凝集抑制劑等,視病情需要給藥;無效時試行血漿置換療法(plasmapheresis)。
飲食保健
以清淡為宜,少量多餐,花樣多變,高熱量、高蛋白、高維生素易消化無渣流質飲食,少食辛辣刺激性的食物。在口腔潰瘍期給予流質飲食,如牛奶、肉湯等,避免過硬、過熱及刺激性食物,以免損傷或加重口腔潰瘍而引起疼痛,應鼓勵患者少量慢食,增加營養,增強體質,促進恢復。

