
簡介
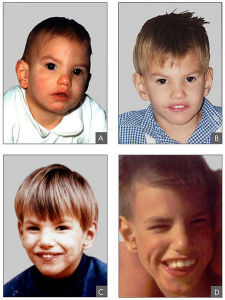 貓哭綜合症病患者的面部特徵︰8 個月大(A), 2 歲(B), 4 歲(C) and 9 歲 6/12 (D)
貓哭綜合症病患者的面部特徵︰8 個月大(A), 2 歲(B), 4 歲(C) and 9 歲 6/12 (D)貓叫綜合症(Cri—du—chat syndrome,CDCS)是由於5號染色體短臂部分缺失所引起的一種染色體病,因在嬰兒期和幼兒期哭聲如貓叫而得名,又稱5p—綜合徵(5p—syndrome)該病較為罕見,須經染色體檢查確診。
1963年,法國科學家首先報告了一種特殊的疾病,病孩的哭聲好像貓在叫一樣,稱為“貓叫綜合徵”。根據國外報告,“貓叫綜合徵”在新生兒中的發病率為四萬五千分之一;在精神發育不全者中,其發生率為3/2000。貓叫綜合症”患兒一般狀態及反應差,哭聲細弱似貓叫樣,頭小而圓,兩眼眶距離過寬,下頜小、頸偏短,耳廓低位,雙手呈“斷掌”掌紋。詢問媽媽獲悉:母親孕早期有先兆流產史。新生兒期餵養困難,吐奶明顯,有黃疸遷延史,出生到現在一直喜哭吵,易激惹,哭聲低微。
該病隨著年齡增長,貓叫樣哭聲好轉。該病死亡率低,多數患兒可活到成人,但體重及身長均低於正常。由於該病有嚴重智慧型障礙及運動發育落後,建議行康復訓練以促進運動功能的發育及智力發育。
核型與遺傳
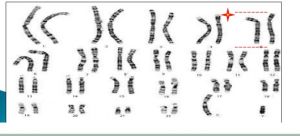 染色體短臂缺失片段
染色體短臂缺失片段患者5號染色體短臂缺失的片段大小不一,經多個DNA探針檢測,證實5p15為本病的缺失片段。80%的病例為染色體片段單純缺失,10%為不平衡易位引起,環狀染色體和嵌合體則比較少見。
病徵與病狀
因為嬰兒的喉頭和神經系統發生問題,所以患有此症的嬰兒會發生貓啼般的哭聲,大約三分之一的小朋友在兩歲之後就不會再發出這種哭聲,除此之外,貓哭症還有以下症狀﹕
1、由於吞咽和吸吮困難而引起餵食問題
2、出生體重低,生長遲緩
3、嚴重的認知、語言和運動發展遲緩
4、行為問題包括:過度活躍症、侵略、暴怒、重複動作
5、隨時間改變的特有面部特徵
6、嚴重流口水
7、便秘
其他常見症狀包括低肌肉張力、小腦症、發育不良、圓臉、脹面頰、小下巴、兩眼過份分離、內眥贅皮、上眼線下鈄、斜視、扁鼻樑、嘴角下垂、低耳、短手指、斷掌、先天性心臟病﹙例如﹕心室間隔缺損、心房間隔缺損、動脈導管未閉、法樂氏四聯症﹚,但患有貓哭症的人,其生育能力則不受影響。
較少見的症狀包括兔唇、顎裂、耳邊有瘺管、胸腺發育不良、腸道轉位不全、巨結腸、腹股溝疝、髖關節脫臼、隱睪症、尿道下裂、罕見腎臟畸形﹙例如﹕馬蹄形腎髒、腎臟異位、發育不全、腎積水﹚、尾指內彎、馬蹄形內翻足、扁平足、第二及第三隻手指和腳趾連趾、可過度伸展的關節等
在較年長的小朋友和青少年的身上,患者會表現出明顯的智力不全、小腦症、臉部特徵變得粗魯、突出的眼眉骨、深陷的眼睛、扁鼻樑、嚴重的咬合不正、脊椎側彎。
受影響的女性患者會踏入青春期,第二性徵、月經會如常出現,生殖器官正常,但曾發現心型的子宮。受影響男性的睪丸通常會比較細小,但可正常製造精子。
診斷依據
 “貓叫綜合徵”是第5號染色體短臂缺失引起的遺傳病
“貓叫綜合徵”是第5號染色體短臂缺失引起的遺傳病1.患兒出生後有貓叫樣哭聲,隨年齡增長而漸消失,2歲後不再存在。女性患者多於男性。
2.小頭、枕部扁平,圓臉,眼距寬,耳位低,下頷小,齶弓高,內眥贅皮,眼瞼外側向下斜。肌張力低或增強,或有耳道、脊椎及四肢的畸形。陰莖、睪丸小。少數合併先天性心臟病。
3.手紋明顯異常。
4.嚴重的智力低下,智商多在25%以下。
5.染色體核型:可有單純5號染色體短臂缺失,5號染色與其他染色體易位,或環形5號染色體等。
發生原因
染色體斷裂的原因
 貓叫綜合症
貓叫綜合症許多環境因素能夠引起染色體斷裂。接觸大劑量粘合劑、油漆;一定劑量的X—射線,一般說來,這種斷裂的出現是暫時性的,經過一定時間可以恢復,不會引起遺傳問題。然而,在懷孕前後有可能對胎兒造成危害。另外病毒感染也是一個嚴重問題。
染色體結構變異
染色體結構變異包括缺失、重複、倒位和易位四種類型。染色體結構變異最早是在果蠅中發現的。遺傳學家們在1917年發現染色體缺失,1919年發現染色體重複,1923年發現染色體易位,1926年發現染色體倒位。人們在果蠅幼蟲唾腺染色體上,對各種染色體結構變異進行了詳細的遺傳學研究。
染色體結構變異的發生是內因和外因共同作用的結果,外因有各種射線、化學藥劑、溫度的劇變等,內因有生物體內代謝過程的失調、衰老等。在這些因素的作用下,染色體可能發生斷裂,斷裂端具有癒合與重接的能力。當染色體在不同區段發生斷裂後,在同一條染色體內或不同的染色體之間以不同的方式重接時,就會導致各種結構變異的出現。下面分別介紹這幾種結構變異的情況。
缺失
缺失是指染色體上某一區段及其帶有的基因一起丟失,從而引起變異的現象。如果缺失的區段發生在染色體兩臂的內部,稱為中間缺失。如果缺失的區段在染色體的一端,則稱為頂端缺失。在缺失雜合體中,由於缺失的染色體不能和它的正常同源染色體完全相應地配對,所以當同源染色體聯會時,可以看到正常的一條染色體多出了一段(頂端缺失),或者形成一個拱形的結構(中間缺失),這條正常染色體上多出的一段或者一個結,正是缺失染色體上相應失去的部分。缺失引起的遺傳效應隨著缺失片段大小和細胞所處發育時期的不同而不同。在個體發育中,缺失發生得越早,影響越大,缺失的片段越大,對個體的影響也越嚴重,重則引起個體死亡,輕則影響個體的生活力。在人類遺傳中,染色體缺失常會引起較嚴重的遺傳性疾病,如貓叫綜合症等
重複
染色體上增加了相同的某個區段而引起變異的現象,叫做重複。在重複雜合體中,當同源染色體聯會時,發生重複的染色體的重複區段形成一個拱形結構,或者比正常染色體多出一段。重複引起的遺傳效應比缺失的小。但是如果重複的部分太大,也會影響個體的生活力,甚至引起個體死亡。例如,果蠅由正常的卵圓形眼變為棒狀眼的變異,就是X染色體上某一區段重複的結果。
重複對生物的進化有重要作用。這是因為“多餘”的基因可能向多個方向突變;而不致於損害細胞和個體的正常機能。突變的最終結果,有可能使“多餘”的基因成為一個能執行新功能的新基因,從而為生物適應新環境提供了機會。因此,在遺傳學上往往把重複看作是新基因的一個重要來源。
倒位
染色體在兩個點發生斷裂後,產生三個區段,中間的區段發生180°的倒轉,與另外兩個區段重新接合而引起變異的現象,叫做倒位。倒位雜合體形成的配子大多是異常的,從而影響了個體的育性。倒位純合體通常也不能和原種個體間進行有性生殖,但是這樣形成的生殖隔離,為新物種的進化提供了有利條件。例如,普通果蠅的第3號染色體上有三個基因按猩紅眼—桃色眼—三角翅脈的順序排列(St—P—Dl);同是這三個基因,在另一種果蠅中的順序是St—Dl—P,僅僅這一倒位的差異便構成了兩個物種之間的差別。
易位
易位是指一個染色體的某一片段移接到另一個非同源染色體上,從而引起變異的現象。如果兩個非同源染色體之間相互交換片段,叫做相互易位,這種易位比較常見。相互易位的遺傳效應主要是產生部分異常的配子,使配子的育性降低或產生有遺傳病的後代。例如,慢性粒細胞白血病,就是由人的第22號染色體和第14號染色體易位造成的。易位在生物進化中具有重要作用。例如,在17個科的29個屬的種子植物中,都有易位產生的變異類型,直果曼陀羅的近100個變種,就是不同染色體易位的結果。
治療
目前尚無有效治療該病的方法,雖發病率低,但對家庭及社會造成的負擔沉重。如何於產前篩查出5p—綜合證胎兒。產前染色體檢查非常重要。
實驗室檢查
1、外周血細胞染色體核型分析。
2、羊水細胞染色體檢查;在孕婦妊娠中期抽取羊水,經細胞培養後作胎兒染色體核型分析。
3、螢光原位雜交。
4、其它輔助檢查:可常規做X線片、超聲心電圖、腦電圖等檢查,部分患兒可發現先天性心臟病,腦電圖異常改變。X線檢查可發現脊柱側彎,並指趾和肋骨畸型等。
優生優育
1、貓叫綜合徵患者不僅能活到成年,且可有生育能力而將缺失染色體傳給子女,使其患同樣的疾病。
2、缺少染色體既可來自新突變或父母的平衡重排,也可直接來自貓叫綜合徵患者。
3、早產、出生體重輕也是該綜合徵患者的重要臨床特徵。所以,為了優生優育,這類成年已婚患者不宜生育。
護理方法
對於貓叫綜合症的嬰兒父母應該精心照顧:
飲食,是孩子成長過程中非常重要的環節,是所有其他生命活動的基礎。要使孩子吃得好,長得健壯,就要先了解孩子的生理特點。中醫認為孩子屬“稚陰稚陽”之體,即孩子體內精血津液還不充實,內臟功能尚不健全或全而未壯。鑒於以上的生理特點,孩子的護養重在調理脾胃。調理脾胃的根本不在於吃什麼靈丹妙藥,而完全在於日常的飲食。那么,日常飲食該如何調養呢?貓叫綜合症的嬰兒的護理八大法則:
一、食貴有節
古人認為,“若要小兒安,須受三分飢與寒”。在貓叫綜合症的嬰兒孩子的飲食方面,既要供應充足的營養,滿足機體生長發育的需要,又要適度適量。如攝食過量,則不僅可能導致營養過剩,還有可能傷及脾胃,導致消化、吸收的障礙。
二、食宜清淡,不可偏食宜食清淡且易於消化的食物。如果胃腸中積累大量不能及時消化的食物,則易產生積滯不化的病理停滯,進而會變化成痰,積久而化熱,痰熱在體記憶體在便成為致病的因素。另外,對貓叫綜合症的嬰兒孩子偏食應該給予足夠的重視,因為偏食必然使營養供給不全面:某些營養過剩,而某些營養又缺乏。這樣會使得營養失衡,導致疾病而影響孩子的生長發育。
三、食宜暖
貓叫綜合症的嬰兒孩子的脾胃功能還在發展之中,胃喜暖而惡濕寒,要保護好脾胃,就要在食物上注意保暖,少吃甜冷冰凍的食品和涼的飯菜。
四、食宜細緩,不可粗速
就餐速度和消化、吸收有密切的關係,因此,吃飯要養成細嚼慢咽的好習慣,以使食物在口中停留的時間長一些,食物能被切磨得碎爛一些,唾液發揮的作用也更充分些。貓叫綜合症的嬰兒孩子吃飯時若狼吞虎咽,易造成胃腸功能紊亂、消化不良,甚至發展成慢性營養障礙性疾病。
五、食前忌動,食後忌靜
一般來說,人在運動時,血液大多集中在肢體、肌肉和除胃以外的其他臟器中,所以在飯前最好不要做劇烈活動,以免胃腸部位因血液缺乏而影響消化。但在吃完飯後,如果呆坐不動,胃腸也會被迫減緩活動量而造成食物停滯在胃腸形成積滯。飯後適當走一走,可以幫助胃腸消化吸收,所以有“飯後百步走,活到九十九”之說。
六、胃好恬愉
情緒會影響飲食消化。當生氣、著急、悲哀、害怕或受驚時,有人會發生噁心、想吐、不想吃飯的現象,這就是情緒影響著消化功能的表現。而進食前情緒特別好時就會吃得很香也很多,吃完後也會很容易消化。所以在飯前、飯後,父母不要批評甚至打罵孩子,否則會使孩子的脾胃受到損傷。
七、食貴有時
主要是說要堅持好一日三餐,要定時定量。如此一來,貓叫綜合症的嬰兒孩子在一定的時間會產生飢餓感,胃腸內會產生大量的消化液,而使吃進的食物能順利地消化和被吸收。
八、慎用醫藥
用藥不當會傷及孩子的元氣。孩子生病要在醫生的指導下用藥,能用天然藥物就不用化學藥物;能用某種物理治療方法就可以達到治療目的的,就不用藥物治療;更不要一味求療效,給孩子吃各種各樣的藥。另外,孩子進補宜慎重。現今生活富足,孩子體質偏虛的少,偏實的多。父母如果不辨明孩子當前的體質情況,一味讓其進補,結果常常是不但無益反而有害,所以尤需慎重。孩子飲食調養的責任在父母,父母應該精心觀察孩子的食慾、精神狀態、睡眠和大小便等狀況,發現異常及時調整。孩子在青春發育期以前,他們的行為、能力都還處在發展階段,還沒有成熟,在吃、喝的問題上不能任意而為,而應在父母的指導下吃和喝。因此,父母掌握好孩子的飲食尤為重要。

