
俗稱
藍眼寶寶
藍眼珠只是瓦登伯革氏症候群諸多症狀當中的一個,患者的眼珠雖呈藍色,視力卻完全不受影響,需要注意的反倒是容易合併聽障及長期便秘的問題,必須早期發現及矯治,以使他們得到良好的療效,健康正常的成長。
臨床症狀
1、藍眼珠或兩眼一藍一正常,稱為虹膜異色症(heterochromia irides) 。 不過也有部分患者 眼珠顏色正常。
2、單耳或雙耳聽力障礙,發生率為 9~38%
3、額前一撮白髮或易有少年白。
4、兩眼眼距較寬,但瞳孔間距離正常,又稱為內眥外移 (dystopiacanthorum ) 。
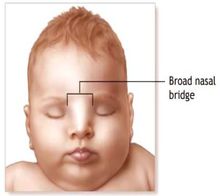
5、鼻根寬闊且鼻翼發育不良。
6、並眉。
7、下巴較大、較寬。
8、長期便秘或甚至同時罹患了先天性巨結腸症。
9、少數有皮膚脫色斑、唇顎裂、先天性心臟病或肌肉、骨骼異常。
 瓦登伯革氏症候群
瓦登伯革氏症候群並不是每位瓦氏症的患者都會有以上的症狀,深入去研究發現可以將此症細分為四型:第一型為典型的瓦氏症,具有上述(一)至(六)的特徵;第二型則為剔除了第一型的內眥外移一項特徵;第三型為第一型的特徵另外再合併有顏面、肌肉或骨骼的異常;第四型則為第一型或第二型合併先天性巨結腸症。此症的發生率約為兩萬至四萬人中有一人,其遺傳型式於第一至第三型屬自體顯性遺傳,偶有新的突變發生,而第四型則屬自體隱性遺傳方式。男女性均可能罹病。瓦氏症是造成聽障的重要原因之一,聽障學童中有1.43%~2.7%患有此症,都是源自於耳蝸的神經性聽力缺陷。
此症的病因目前已有較明確的答桉,可能是胚胎時期神經嵴衍生細胞無法正常移行分化所導致的結果,而黑色素細胞、聽覺神經細胞及結腸神經結細胞均源自神經嵴細胞。整個的分化移行過程頗為複雜,需要PAX3、MITF、EDN3、EDNRB及SOX 10等基因參與協同運作,因此任何一個基因有了缺陷,均會造成瓦氏症,而有臨床上形形色色大同中卻有小異的症狀。分子生物學的研究在近年內有長足的進步,許多各種型式的基因突變被發現及解讀,日後將對此症致病機轉的闡明、臨床遺傳診斷的套用與基因療法的發展產生重大的影響。
遺傳型式
發生率約是兩萬分之一到四萬分之一其遺傳型式於第一至第三型屬體染色體顯性遺傳 ,不分性別,每胎均有1/2 機率可罹患此症,偶有新的突變發生,而第四型則屬體染色體隱性遺傳方式,不分性別,每胎均有1/4 機率可罹患此症。
遺傳變異自這項疾病被提出後,陸續有不少學者提出它的變異表現型,可歸類為四型:
| | 相關基因//染色體位置 | 症狀 |
| 第一型典型瓦氏症 | PAX3 genechromosome 2 | 內眼外移易位、鼻根寬闊、並眉、額前白髮、完全或是部分紅膜異色、先天性一側或是兩側聽障(25%)六項主要特徵 |
| 第二型 | MITF genechromosome 3 | 除內眼移位,但仍包括其他五項特徵聽障(55%) |
| 第三型 | PAX3 genechromosome 2 | 為第一型的特徵……另外再合併有顏面、肌肉或骨骼的異常與發育不全。有些還有小頭症及重度智障 |
| 第四型 | EDNRB 與EDN3 genes | 第一型或第二型合併先天性巨結腸症先天性心臟疾病 |
致病機轉
此症的病因目前已有較明確的答桉,可能是 胚胎時期神經嵴衍生細胞無法正常移行分化 所導致的結果,而黑色素細胞、聽覺神經細胞及結腸神經結細胞均源自神經嵴細胞。
整個的分化移行過程頗為複雜,需要 PAX3 、MITF 、EDN3 、EDNRB 及SOX 10 等基因參與協同運作,因此任何一個基因有了缺陷,均會造成瓦氏症候群,而有臨床上形形色色大同中卻有小異的症狀。
預防及篩檢
現階段瓦氏症患者及家庭最需要注意的事項是,當家族中有藍眼珠的成員,則所有同血源的親屬 均應接受遺傳專科醫師的檢查,以查出可能的病者 (瓦氏症患者不一定都有藍眼珠),進一步接受聽力篩檢,必要時須查驗有無巨結腸症或心臟血管系統、肌肉骨骼系統的病變,以便及早給予適當的治療及復健。
治療
目前沒有有效治療
聽力障礙的治療與復健:
瓦氏症候群所致的聽障,其機率約在 50% 左右,多數是 感覺神經性聽障 ,且其程度不等。一般最好在一歲以前,就必須予以確認。首先必須予以孩子 配帶合適的助聽器 ,以期儘早刺激腦部之聽覺中樞 (一般腦的可塑性在三歲以前是最好的 )。若雙耳之聽力仍在 60 分貝以下,必須考慮手術植入“人工電子耳” ,再加上長時間的聽語復健。
眼睛照顧:
藍眼珠的小朋友,因為色素比較少,自然對較強的光線會畏懼 ,可以戴有帽緣的帽子或上太陽眼鏡避光或補充人工淚水也可以 。

