
病因
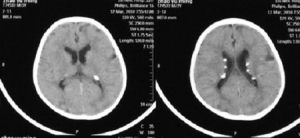 結節性硬化症CT影像
結節性硬化症CT影像遺傳方式為常染色體顯性遺傳,家族性病例約占三分之一,即由父母一方遺傳而來突變的TSC1或TSC2基因;散發病例約占三分之二,即出生時患者攜帶新突變的TSC1或TSC2基因,並無家族成員患病。家族性患者TSC1突變較為多見,而散發性患者TSC2突變較常見。
臨床表現
根據受累部位不同,可有不同表現。典型表現為面部皮脂腺瘤、癲癇發作和智慧型減退。多於兒童期發病,男多於女 。1.皮膚損害
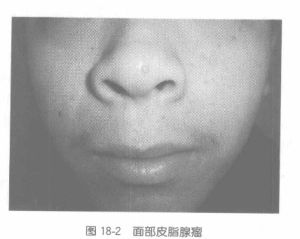 臨床表現
臨床表現2.神經系統損害
(1)癲癇為本病的主要神經症狀,發病率占70%~90%,早自嬰幼兒期開始,發作形式多樣,可自嬰兒痙攣症開始,至部分性局灶性或複雜性發作、全面性大發作。頻繁而持續的癲癇發作後可繼發違拗、固執等癲癇性人格障礙。若伴有皮膚色素脫失可診斷為結節性硬化症,以後轉化為全面性、簡單部分性和複雜部分性發作,頻繁發作者多有性格改變。
(2)智慧型減退多進行性加重,伴有情緒不穩、行為幼稚、易衝動和思維紊亂等精神症狀,智慧型減退者幾乎都有癲癇發作,早發癲癇者易出現智慧型減退,癲癇發作伴高峰節律異常腦電圖者常有嚴重的智慧型障礙,部分患者可表現為孤獨症。
(3)少數可有神經系統陽性體徵。如錐體外系體徵或單癱、偏癱、截癱、腱反射亢進等,如室管膜下結節阻塞腦脊液循環通路或局部巨大結節、並發腫瘤等可引起顱內壓增高表現。
3.眼部症狀
50%患者有視網膜膠質瘤,稱為晶體瘤。眼底檢查在眼球後極視乳頭或附近可見多個蟲卵樣或桑椹樣鈣化結節,或在視網膜周邊有黃白色環狀損害。此外尚可出現小眼球、突眼、青光眼、晶體混濁、白內障、玻璃體出血、色素性視網膜炎、視網膜出血和原發性視神經萎縮。
4.腎臟病變
腎血管平滑肌脂肪瘤(AML)和腎囊腫最常見,表現為無痛性血尿、蛋白尿、高血壓或腹部包塊等,在TSC死亡者中因腎臟疾病而夭折者約占27.5%,是該病死亡的第二大原因。
5.心臟病變
47%~67%患者可出現心臟橫紋肌瘤,該腫瘤一般在新生兒期最大,隨年齡增大而縮小至消失,可引起心力衰竭,是本病嬰兒期最重要的死亡原因,產前超聲最早能在妊娠22周時發現腫瘤,提示患TSC的可能為50%。
6.肺部病變
肺淋巴管肌瘤病(LAM)累及肺部常見於育齡女性患者,是結締組織、平滑肌及血管過度生長形成網狀結節與多發性小囊性變,可出現氣短、咳嗽等肺心病、自發性氣胸的表現。
7.骨骼病變
骨質硬化,顱骨硬化症最為常見,亦好發於指、趾骨,為骨小梁增生所致;囊性變,全身骨骼均可受累,X線可發現;脊柱裂和多趾(指)畸形。
8.其他臟器
包括消化道、甲狀腺、甲狀旁腺、子宮、膀胱、腎上腺、乳腺、胸腺等均可能有受累,目前認為TSC除骨骼肌、松果體外可累及所有組織器官。
檢查
1.頭顱平片腦內結節性鈣化和因巨腦回而導致的巨腦回壓跡。
2.頭顱CT或MRI
平掃可見室管膜下腦室邊緣及大腦皮層表面多個結節狀稍低或等密度病灶,部分結節可顯示高密度鈣化,為雙側多發性,增強呈普遍增強,結節更清晰,可發現平掃不能顯示的結節。皮層和小腦的結節有確診意義。
3.腦電圖
可見高幅失律和各種癲癇波。
4.腦脊液
正常。
5.腹部超聲
可見腎血管平滑肌脂肪瘤、腎囊腫、多囊腎。
6.超聲心動圖
新生兒及嬰幼兒易發現心臟橫紋肌瘤,腫瘤在最初三年內變小過程顯著,成年逐步消失,故大齡兒童及成人檢測陽性率低。
7.心電圖
可發現心律失常,常見預激綜合徵。
8.胸部X線
可發現肺部錯構瘤、氣胸等該病。
診斷
1.確診的TSC2個主要指征或一個主要指征加上兩個次要指征;
2.擬診的TSC
1個主要指征加上1個次要指征;
3.可能的TSC
1個主要指征或2個及以上次要指征。
4.主要指征
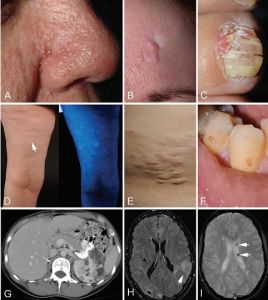 成年女性結節性硬化症患者的病情特點
成年女性結節性硬化症患者的病情特點(2)非外傷性指(趾)甲或甲周纖維瘤;
(3)色素減退斑(≧3);
(4)鯊革樣皮疹(結締組織痣);
(5)多發性視網膜錯構瘤結節;
(6)皮質結節;
(7)室管膜下結節;
(8)室管膜下巨細胞星形細胞瘤;
(9)單個或多發的心臟橫紋肌瘤;
(10)肺淋巴管性肌瘤病;
(11)腎血管平滑肌瘤。
5.次要指征
(1)多發性、隨機分布的牙釉質凹陷;
(2)錯構瘤性直腸息肉(組織學證實);
(3)骨囊腫(放射學證實);
(4)腦白質放射狀移行束(放射學證實);
(5)牙齦纖維瘤;
(6)非腎性錯構瘤(組織學證實);
(7)視網膜色素缺失斑;
(8)Confetti皮損;
(9)多發性腎囊腫(組織學證實)。
6.備註
(1)若腦內皮質發育異常與腦白質移形束同時存在,只能算一個指征;
(2)若肺淋巴管性肌瘤病與腎血管平滑肌瘤共存,則需有其他TSC指徵才能確診;
(3)腦白質移形束與局灶皮質發育異常常見於TSC患者,但因其常單獨出現且不具特異性,故只做為次要指征。
鑑別診斷
根據其多系統、多器官受累特點,需與其他累及皮膚、神經系統和眼的疾病鑑別,如神經纖維瘤病和腦面血管瘤病。在0.8%的新生兒中可發現色素減退斑,大多數並無醫學意義,其他一些疾病如白癜風、色素缺失痣部分白斑病及Vogt-Koyanagi-Harade綜合徵也可有色素減退斑,需與TSC鑑別。有癲癇表現的患者需與原發或繼發性癲癇鑑別。影像學上需與腦囊蟲病鑑別。治療
由於TSC1和TSC2蛋白參與調節哺乳動物雷帕黴素靶蛋白(mTOR)激酶活性,因此考慮套用雷帕黴素治療TSC。雷帕黴素屬於大環內酯類抗生素,因抑制mTOR活性、參與調節細胞生長用於抗真菌治療,在器官移植術後也作為免疫調節藥物套用。抗癲癇治療,早期控制癲癇發作有助於預防繼發的癲癇腦病和認知行為損害,嬰兒痙攣症的治療推薦個體化用藥方案,研究顯示氨己烯酸對73%的TSC嬰兒痙攣症患者有效,TSC相關癲癇可能對多種抗癲癇藥效果不佳,部分小樣本研究顯示外科手術治療可達到滿意效果。
腎血管平滑肌脂肪瘤,腫瘤直徑大於3.5cm~4.0cm時易發生出血而出現疼痛,此時有干預指征,可行腎動脈栓塞或腎部分切除術。部分研究也證實mTOR抑制劑對腎血管平滑肌脂肪瘤有效,但FDA尚未批准此適應證。
肺淋巴管肌瘤病多見於育齡婦女,提示雌激素可能參與刺激肺部平滑肌細胞生長。孕酮激素治療和/或卵巢切除術可減少雌激素產生,但治療效果個體差異較大。也有研究顯示mTOR抑制劑對肺淋巴管肌瘤病有效,同樣FDA也尚未批准此適應證。
其他對症治療包括脫水降顱壓,腦脊液循環受阻可行手術治療,面部皮脂腺瘤可整容治療。
飲食保健
宜清淡為主,多吃蔬果,合理搭配膳食,注意營養充足,謹遵醫囑。預防護理
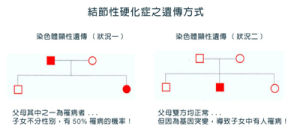 遺傳方式
遺傳方式
