
簡介
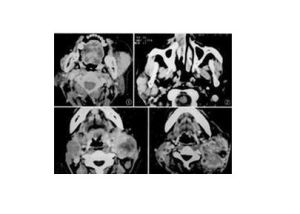 神經母細胞瘤
神經母細胞瘤神經母細胞瘤是起源於胚胎性交感神經系統神經嵴細胞的惡性腫瘤,可分泌各種神經原性衍生物(兒茶酚胺、神經元特異烯醇酶等)。發病率占小兒惡性實體瘤的第三位,多見於5歲以下小兒,也可見年長兒發病者。病情進展快,轉移發生早,預後較差。1歲以內患兒的腫瘤有自發消退傾向。
病因
發病原因
屬胚胎性腫瘤,多位於大腦半球。
發病機制
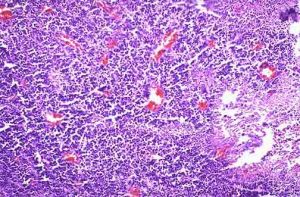 顯微鏡下,神經母細胞瘤是一個“小的蘭圓細胞”腫瘤。組織學變異,所處階段和細胞遺傳學特點幫助決定預後。
顯微鏡下,神經母細胞瘤是一個“小的蘭圓細胞”腫瘤。組織學變異,所處階段和細胞遺傳學特點幫助決定預後。 NB來自起源於神經嵴的原始多能交感神經細胞,形態為藍色小圓細胞。從神經嵴移行後細胞的分化程度、類型及移行部位形成不同的交感神經系統正常組織,包括脊髓交感神經節、腎上腺嗜鉻細胞。NB組織學亞型與交感神經系統的正常分化模型相一致。經典的病理分類將NB分成3型,即神經母細胞瘤、神經節母細胞瘤、神經節細胞瘤,這3個類型反應了NB的分化、成熟過程。典型的NB由一致的小細胞組成,約15%~50%的病例,母細胞周圍有嗜酸性神經纖維網。另一種完全分化的、良性NB為神經節細胞瘤,由成熟的節細胞、神經纖維網及Schwann細胞組成。神經節母細胞瘤介於前兩者之間,含有神經母細胞和節細胞混雜成分。
Shimada分類結合年齡將病理分成4個亞型,臨床分成2組。4個亞型即包括NB(Schwannin少基質型);GNB混合型(基質豐富型);GN成熟型和(3NB結節型(包括少基質型和基質豐富型)。前3型代表了NB的成熟過程,而最後一型則為多克隆性。對NB而言,細胞分化分為3級,包括未分化、分化不良、分化型;細胞的有絲分裂指數(MKI)也分為低、中、高3級。Shimada分類綜合腫瘤細胞的分化程度、有絲分裂指數和年齡,將NB分為臨床預後良好組(FH)和預後不良組(UFH):
1.FH包括以下各類
(1)NB,MKI為低中度,年齡<1.5歲。
(2)分化型NB,MKI為低度,年齡1.5~5歲。
(3)GNB混合型。
(4)GN。
2.UFH包括
(1)NB,MKI高級。
(2)NB,MKI為中級,年齡1.5~5歲。
(3)未分化或分化不良型NB,年齡1.5~5歲。
(4)所有>5歲的NB。
(5)GNB結節型。
在病理上,除HE染色外,可進一步做免疫組化電鏡檢查來與其他小圓細胞腫瘤相鑑別,NB時神經特異性酯酶(NSE)陽性,電鏡下可見典型的緻密核,結合於膜上的神經分泌顆粒,在神經纖維網中有微絲和平行排列的微管。
臨床表現
臨床表現與原發部位、年齡及分期相關。65%患兒腫瘤原發於腹腔,大年齡兒童中腎上腺原發占40%,而在嬰兒中只占25%。其他常見部位為胸腔和頸部。約10%病例原發部位不明確。約70%NB在5歲前發病,極少數在10歲以後發病。
不同部位的腫塊
最常見的症狀為不同部位的腫塊。
(1)原發於腹部:以腎上腺及脊柱二側交感神經鏈原發多見,一般在腫塊較大時才出現症狀,可有腹痛、腹圍增大、腰背部飽滿、捫及腫塊、胃腸道症狀。
(2)原發於胸腔:有縱隔壓迫相關症狀及呼吸道症狀,如氣促、咳嗽等。
晚期表現
病人常有肢體疼痛、貧血、發熱、消瘦、眼眶部轉移。眼眶部轉移形成具有特徵性的熊貓眼,表現為眼球突出、眶周青紫。其他可有高血壓及腫塊部位相關壓迫症狀,如有椎管內浸潤壓迫時出現運動障礙、大小便失禁等。
轉移途徑
NB主要轉移途徑為淋巴及血行。在局限性病變病人中約35%有局部淋巴結浸潤,血行轉移主要發生於骨髓、骨、肝和皮膚,終末期或復發時可有腦和肺轉移,但較少見。嬰兒病例就診時局限性病變、局限性病變伴有局部淋巴結轉移、播散性病變分別為39%、18%和25%;但在大年齡兒童中分別為19%、13%和68%,也即大年齡患兒就診時多數已處疾病晚期。
診斷
診斷方法
組織病理學檢查是NB診斷的最重要手段,有時需結合免疫組織化學、電鏡以明確診斷。影像學檢查發現有與NB特徵相符合的腫塊,同時骨髓中發現NB腫瘤細胞,有明顯增高的兒茶酚胺代謝產物(HVA或VMA)也可做出診斷。如病理診斷有困難時,染色體檢查發現有1p缺失或N-myc擴增支持NB診斷。
診斷分期
同時應包括診斷分期,美國兒童腫瘤協作組分期系統(CCSG)如下:
(1)Ⅰ期:腫瘤局限於原發器官。
(2)Ⅱ期:腫瘤超出原發器官,但未超過中線,同側淋巴結可能受累。
(3)Ⅲ期:腫瘤超過中線,雙側淋巴結可能受累。
(4)Ⅳ期:遠處轉移。
(5)Ⅵs期:<1歲,原發灶為Ⅰ、Ⅱ期,但有局限於肝、皮膚、骨髓的轉移灶。
檢查
儘量爭取病理活檢以明確診斷及分型。為確定病變範圍及臨床分期,應做骨髓活檢或塗片,骨髓中發現NB腫瘤細胞,85%~90%患兒尿中兒茶酚胺代謝產物同型香酸(HVA)、香草基杏仁酸(VMA)增高。NB時血LDH可升高,並與腫瘤負荷成正比。可用螢光原位雜交法(FISH)檢測腫瘤細胞N-MYC的擴增情況,如大於10倍,常提示預後不良。細胞遺傳學檢查可發現1p-或N-myc擴增。
選擇性骨骼X線平片、X線胸片、骨掃描,胸、腹部CT或MRI。影像學所示腫塊中常有鈣化灶,原發於胸腔時多見於後縱隔脊柱兩側,原發於腹腔時多見於腎上腺或後腹膜脊柱兩側。
治療
由於NB預後差異大,部分病人如小年齡、早期NB預後明顯優於大年齡晚期組,因此應根據病人的預後因素,如年齡、分期、N-myc擴增、1p缺失等採用分級治療。早期病人無N-myc擴增及1p缺失,可僅做手術,手術後隨訪。而大年齡、晚期,伴有N-myc擴增,1p缺失者,需接受強化療和手術,直至骨髓移植。
手術、化療、放療仍為NB治療的三大主要手段,根據其臨床預後因素採用不同強度的治療方案。一般對局限性腫瘤主張先手術切除,再化療。而對估計手術不能切除者採用先化療、再手術、再化療或加放療的策略。對NB敏感的藥物有環磷醯胺、長春新鹼、依託泊苷(VP-16)、卡鉑、順鉑、抗腫瘤抗生素(多柔比星)、異環醯胺等,各個協作組採用不同藥物組合對晚期病人強化療,但預後改善仍未令人滿意。
美國CCSG協作組報導晚期NB在接受自身骨髓移植後4年無進展性疾病生存率為38%,各項處理方案結果未顯示有差別。對Ⅳ期具有其他預後不良因素者(如N-myc擴增,年齡>2歲,誘導治療未獲緩解者),自身骨髓移植組預後要比常規治療好。異基因移植與自體移植間結果無差異。自體外周血幹細胞移植時造血功能恢復要比骨髓幹細胞移植快,並且腫瘤細胞污染的機會相對減少。
NB對放療敏感,但全身放療在幹細胞移植預處理方案中的套用尚有爭論。NB的原發部位復發機會較高,因此對Ⅲ、Ⅳ期病人仍有主張化療同時採用局部放療,但其有效性不明確。全身照光並不改善預後,對晚期疼痛病人,照光可緩解疼痛。
美國兒童腫瘤協作組對晚期病人在自身幹細胞移植後隨機分組進行13-順維A酸治療研究,一組病人接受160mg/(m2·d),每月用2周,共3~6個月,另一組病人停化療後不用藥。結果為接受維A酸組3年EFS為47%,未接受組為25%,P=0.013。在Ⅳ期病人及高危Ⅲ期病人中維A酸作用更為明顯,分別為40%對22%,和77%對49%。一般21~28天為一療程。
預後
分期及年齡
為最重要的預後因素,Ⅰ、Ⅱ期,Ⅵs期預後明顯優於Ⅲ、Ⅳ期。<1歲者明顯優於>2歲者。晚期大年齡患兒的長期無病生存率僅為5%~30%。
生物學特徵
在NB中常見有N-myc擴增,N-myc對細胞分裂有正向調節作用,維A酸(RA)對N-myc表達有負向調節作用以致NB細胞停止增殖並分化,N-myc擴增>10倍為預後不良因素。1p36.3缺失是易復發的因素,1p可能有腫瘤抑制因子,即使無N-myc擴增,1p36.3缺失仍有意義。17q獲得(gain)時預後差。NB表達酪胺酸激酶(Trk)家族受體激酶的研究進展較快,預後良好型表達TrkA、C;而預後不良型、N-myc擴增型表達TrkB。CD44是一種黏附分子,在NB中CD44的表達與NB進展之間的關係恰與其他腫瘤相反,CD44陽性無病生存率顯著高於CD44陰性組,CD44的表達與N-myc倍增呈負相關。
病理型別
Shimada分類中UFH預後差。
併發症
可發生貧血、消瘦高血壓、運動障礙可發生轉移。
治療方案
治療方案取決於病情。不同來源的資料對治療方法的描述也不盡相同。以下說法僅供參考(摘自“小兒腫瘤天地”網站)。
1期患兒完整切除原發腫瘤後無需進一步治療。近年強調1歲以下的1期腫瘤多可自然消退,主張可密切隨訪,暫不手術。
2期患兒對組織結構良好、無淋巴結轉移、NSE和鐵蛋白正常,N-myc基因拷貝數<10和DNA異倍體的病例,完整切除原發腫瘤後可不予其它治療;而對組織結構不良、淋巴結陽性、腫瘤標記物(NSE、鐵蛋白)數值升高,DNA二倍體,N-myc拷貝數>10,手術切除後應常規化療12個月,必要時還需局部放療。
3期病例腫瘤完全切除者,根據組織結構、淋巴結浸潤、腫瘤標記物、N-myc基因擴增、DNA倍體檢測結果,決定放療劑量(15-30Gy)和術後化療時間(12-18個月)。而腫瘤未完全切除,術後化療3-6個月後仍有腫瘤殘留或腫瘤標記物(VMA、HVA、NSE、鐵蛋白)高於正常或淋巴結增大,應予二次手術或二次探查,常規區域淋巴結清掃,腫瘤床剝除,術後化療18個月。腫瘤巨大判斷不能切除者,應術前化療後再予延期手術。

