
疾病介紹
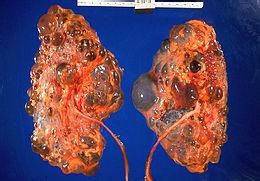 常染色體顯性多囊腎
常染色體顯性多囊腎發病率約為1/1000,其外顯率近乎完全,這使得所有活到80歲以上的攜帶者均顯示出本病的某些徵象。約5%~10%終末期腎衰是由常染色體顯性多囊腎(ADPKD)導致。
常染色體顯性多囊腎是由什麼原因引起的?
本症確切病因尚不清楚。儘管大多在成人以後才出現症狀,但在胎兒期即開始形成。囊腫起源於腎小管,其液體性質隨起源部位不同而不同,起源於近端小管,囊腫液內成分如Na+、K+、CI-、H+、肌酐、尿素等與血漿內相似;起源於遠端則囊液內Na+、CI-濃度較低,而K+、H+、肌酐、尿素等濃度較高。
大多數患者的異常基因位於16號染色體的短臂,稱為ADPKD1基因,基因產物尚不清楚。少數患者的異常基因位於4號染色體的短臂,稱為ADPKD2基因,其編碼產物也不清楚。兩組在起病、高血壓出現以及進入腎功能衰竭期的年齡有所不同。
常染色體顯性多囊腎有哪些表現及如何診斷?
晚期病例腎臟明顯腫大可捫及容易診斷。尿液分析見輕度蛋白尿和不同程度的血尿,但紅細胞管型不常見。即使並沒有細菌感染,膿尿症多見。由於囊腫破裂或結石移動也可有發作性的明顯肉眼血尿。靜脈尿路造影檢查具有特徵性,表現為有多個囊腫,及由此引起的腎臟腫大,外形不規則,並且因為囊腫壓迫腎盞,漏斗和腎盂而使其拉長呈蜘蛛狀。由於囊腫取代功能性組織,故在肝,腎的超聲檢查和CT掃描中可顯示典型的"蟲蝕"狀。因此在靜脈尿路造影未顯示典型改變之前,這些檢查可作為該病早期診斷的手段。需與該病相鑑別的是尚未造成足夠腎實質損害導致尿毒症的單個或多發性囊腫。
正確的遺傳學診斷很快可採用。運用重組DNA技術已發現約85%的APD-KD家族中,被稱為PKD1的基因突變定位於16號染色體短臂(P)上,它具有兩個特異性標誌:α球蛋白複合體及磷酸甘油酸激酶的基因。大多數其餘的家族發現在4號染色體(PKD2)上有基因缺陷,但也有少數家族不與如何基因座相關。
常染色體顯性多囊腎應該做哪些檢查?
影像學檢查,包括超音波、CT及磁共振等。
常染色體顯性多囊腎容易與哪些疾病混淆?
鑑別診斷主要考慮與多房性單純腎囊腫鑑別,其他鑑別需考慮ARPKD,獲得性腎囊腫,多囊性腎發育不良等。表19-30 早期ADPKD與多房性單純性腎囊腫鑑別。
常染色體顯性多囊腎可以並發哪些疾病?
尿路感染最常見,大多為下尿路感染。也可出現腎盂腎炎,囊腫感染等。其他併發症有:尿路結石、梗阻;動脈瘤破裂出血,特別是顱內動脈瘤破裂占ADPKD患者死因的7%~13%。極少見情況下可出現兩腎的惡性腫瘤。
常染色體顯性多囊腎應該如何治療?
約50%的具有ADPKD1突變的患者在55~60歲之間發展到尿毒症。而非ADPKD1突變的要到70歲才發生。少數ADPKD患者在少兒時就出現臨床表現,即使其父母成年後發病。與許多其他腎臟疾病一樣,ADPKD在黑人中有進展加速,約提早10年發病。其他預示該病更快進展的因素包括年幼時即診斷,男性,腎臟體積較大,高血壓,肝囊腫(在女性患者中),肉眼血尿及尿路感染(男性)。如未進行透析或腎移植,患者常死於尿毒症或高血壓併發症,約10%的患者死於動脈瘤破裂引起的顱內出血。
對尿路感染和繼發性高血壓進行有效的治療可明顯延長生命。若尿毒症存在其處理與其他腎臟病相同。ADPKD透析治療患者的血紅蛋白水平高於其他類型的患者。腎移植是可行的,但由於該病的家族特性,使用雙親和同胞供腎臟。推薦遺傳諮詢。
常染色體顯性多囊腎應該如何預防?
與患者年齡,起病年齡,高血壓的控制程度,是否反覆發作尿路感染,血尿等有關。隨著透析及腎移植技術的不斷提高,患者的主要死因為感染、心血管疾患(心梗、心衰等)以及顱內出血。
常染色體顯性多囊腎吃什麼好?
適宜食物:瘦羊肉、田雞
忌吃食物:大蔥、大蒜、姜
1.保持心情舒暢和樂觀向上的情緒,樹立起戰勝疾病的信心。囊腫性疾病是先天和後天各種因素相互作用的結果,科學研究發現,所有這些因素都是可以改變或加以控制消除的,因此,千萬不可悲觀失望,況且樂觀向上的思想情緒可以提高人的免疫力,有利於戰勝疾病。但另一方面也要克服“輕敵”思想,積極地配合醫生進行治療。樂觀向上,認真對待才是正確的指導思想。
2.及時治療,科學用藥

