
簡介
早老症(Hutchinson-GilfordProgeriaSyndrome,HGPS)是一種早發而嚴重的過早老化性疾病。它是由於編碼A/C型核纖層蛋白的LMNA基因發生點突變而引起。這個突變激活了基因11號外顯子上一個隱蔽的剪接位點,產生了一種被截短了50個胺基酸的A型核纖層蛋白。病因病機
 染色體
染色體臨床表現
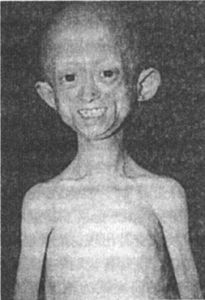 早老症
早老症2、特徵性變化是:
①身長體重明顯低於正常,10歲後,依然如4—5歲的小兒。到18歲,平均身高僅有l17厘米,體重只有16.5公斤。
②皮膚薄而乾燥,有皺紋,並出現棕色老人斑。皮下脂肪逐漸變薄,全身瘦削,面頰的脂層也消失,只剩恥骨上部的皮下脂肪。由於顏面骨及下頜骨特別小,頭顱與前額相對地較大。眼眶較小故兩眼突出,鼻突出且尖;耳郭常有畸形,兩耳向前豎起,缺乏耳垂;嘴唇較薄,近似鳥臉。
③脫髮由枕部開始,至3~5歲時幾乎全部脫光,眉毛與睫毛也可脫落。
④語音尖而細。
⑤乳齒和恆齒均發育延遲,並很早脫落,牙齒畸形。
⑥四肢與軀幹比例正常,鎖骨發育不全,特別短小,關節相對粗大並僵直,手指屈曲,指、趾甲常萎縮,末節指骨很短。
⑦下腹部、大腿近端和臀部硬腫。
⑧頭皮靜脈怒張。
⑨四肢淺表血管粗厚而顯露,尤以橈動脈和手背靜脈最為明顯。
⑩血壓異常,患兒血壓於5歲以後明顯上升,心臟逐漸擴大,有時出現心絞痛、心肌梗死或腦血管意外。也可發生腎結石而致急腹痛。步態如老人,走路時腳拖在地上,不能抬高。
3、北京兒童醫院、北京醫學院兒科和阜外醫院等單位曾觀察1例,自10歲開始,至12歲死亡,屍檢證明全身動脈粥樣變和心肌梗死。曾經試用硫氧嘧啶減少熱量消耗,並長期套用睪酮和蜂王漿,未能增加體重。
4、根據症狀診斷此病不難。有時早期出現口鼻周圍發紺,腹股等處有硬化樣皮膚變化,似可助早診。
輔助檢查
在實驗室檢查方面,可見血清蛋白結合碘、膽固醇、三醯甘油及β脂蛋白皆高於正常值,基礎代謝率也偏高。甲狀腺、腎上腺和垂體功能未見異常。對胰島素有拮抗現象,偶見胰島素依賴性糖尿病。對生長激素反應正常。但性腺不發育或性發育明顯遲緩。亦可見DNA修復降低。1.X線檢查骨骼X線檢查可見輕度脫鈣,骺端肥大及乾骺癒合較早,前囟持久開放,鎖骨小或者由於骨分解而消失,指趾節末端有時不顯影,在2~3歲時往往可見髖外翻。
2.心電圖檢查晚期心電圖顯示冠狀動脈供血不足。
鑑別診斷
1.外胚葉發育不全:是一種遺傳性疾病,缺乏毛髮與指趾甲異常和早老症相似,並可因缺少汗腺而於夏季發生高熱,但無高血壓、消瘦及老人面貌。2.先天性全身性脂肪營養障礙(congenitalgeneralizedlipodystrophy)即Seip-Lawrence綜合徵:是一種原因未明的少見病,在嬰兒早期即見皮下脂肪消失而肌肉、骨骼的生長較快,其面部消瘦,胸、臂亦細小,但下肢正常。往往伴有糖尿病、垂體功能異常等內分泌疾病,屬於常染色隱性遺傳,亦缺治療方法。
3.Cockayne綜合徵:是常染色體隱性遺傳病,可有家族史,其皮下脂肪消失、尖鼻、耳朵畸形、體格矮小等症狀很像早老症,但這些表現常在4歲左右出現,皮膚對日光敏感,且常見小瞳孔、遠視、白內障、視網膜色素沉著、視神經萎縮、神經性耳聾、智力低下,甚至發生粗大震顫和步態蹣跚等,可與早老症區別。
4.werner綜合徵 :treiff綜合徵亦有牙齒缺陷、脫髮、頭髮少及下頜小等畸形,但智力落後,有白內障和小眼畸形,可資鑑別。5.Hallermann-Streiff綜合徵:亦有牙齒缺陷、脫髮、頭髮少及下頜小等畸形,但智力落後,有白內障和小眼畸形,可資鑑別。
6.先天性皮膚鬆弛症:有時須與先天性皮膚鬆弛症鑑別,後者常有皮膚鬆弛現象,不見於早老症。
疾病治療
1、治療尚無特殊療法,可做理療、按摩和堅持體力活動,以減輕運動障礙。一般選用降血脂藥物,並限制多脂飲食,以減輕動脈硬化。套用小劑量阿司匹林(乙醯水楊酸),是否可延緩心肌梗死,尚須進一步觀察。 2、預後 患者大都發生動脈硬化,多於10歲以後死於心肌梗死或腦血管疾患,多數患兒的生存期為7~37歲,平均存活年齡為13.4歲,日本1例報導活到40歲。老人常見的現象如耳聾、遠視、白內障、角膜老人環、關節病以及老年性格改變等多不出現。

