疾病簡介
 基因圖
基因圖臨床症狀
主要症狀通常患者在嬰兒早期的發育正常,但到六個月的時候,血清的磷值開始降低,周歲時嬰兒便出現生長遲緩的現象,但智力發展不受此症影響。患者於開始站立或走路時不願去承受身體的重量。將近1/3患者有聽力問題,成人患者約10%有不同程度之眩暈和噁心經驗。僅零星個案沒有癲癇(seizures)及其它關於肌肉及氧化代謝功能的全身性徵兆。
骨骼肌肉
●於出生第一年的小孩可見關節空間變寬,以及膝蓋磨損的情形。
●下肢彎曲﹑髖關節內彎﹑膝關節內彎或外彎,嚴重者在胸廓肋骨會有佝僂症串珠。
●骨頭軟弱易疼痛﹑肌肉無力易跌倒以及發生自發性骨折。
●輕至重度的發育不良,生長遲緩,成人身高多在130至160公分之間。
牙齒
●齒髓腔大
●琺瑯質發育不全
●乳齒會有長牙遲緩或缺牙的情形
●恆齒周圍多處的自發性感染及膿瘡
併發症
●不成比例的身材矮小
●腎小管酸血症
●假姓副甲狀腺機減退
●低鈣尿症
●次發性的骨骼外結石,例如腎結石
診斷原則
X光攝影術的檢查骨端擴大呈杯狀
骨梁粗大
長骨彎曲
顱骨扁平
脊柱背彎
胸廓肋軟骨變大,即佝僂症串珠
橫切的光可透性線條
生化檢定
檢測血清中磷酸鹽的濃度:嬰幼兒時期的正常濃度範圍為5.0-7.5mg/dL,成人的為2.7-4.5mg/dL。
基因檢驗
目前已知性聯遺傳低磷酸鹽佝僂症是位於X染色體短臂22.1位置上的PHEX基因,此基因表現後可製造出749個胺基酸,鑲嵌於細胞膜上的蛋白酶。PHEX基因發生突變使其的蛋白質產物調節腎臟中磷酸鹽的功能發生缺陷,因而造成磷酸鹽的過度分泌而流失。如欲檢驗此基因是否存在突變,需至門診抽血,經DNA萃取後,再利用分子生物技術做突變分析。已生育患有此症之病童,或患有此症之父母欲再生育下一胎時,若已確定此基因之突變點,可於妊娠第十六周時抽取羊水,經離心後取得胎兒細胞並抽取DNA,與患有此症之父母或兄姊之基因做連鎖分析,已確認胎兒是否遺傳突變基因。
遺傳諮詢
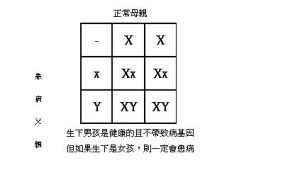
人類細胞共有23對染色體,是由22對體染色體及1對性染色體組成。女性具有22對體染色體及XX的性染色體,男性則有22對體染色體及XY性染色體。性連遺傳低磷酸佝僂症是以性染色體顯性遺傳方式遺傳。顯性遺傳的遺傳模式,只要一對染色體中的一個染色體有病變基因就會導致疾病的發生。此疾病病變的基因位於性染色體的X染色體的短臂22.1的位置上,是個可製造749個胺基酸蛋白質的基因,基因名字是PHEX。有三分之二的性連遺傳低磷酸佝僂症具有家族史,也就是經由遺傳來的。回溯病童的家族史,可發現父親或母親其中一方,經臨床或基因診斷後,確實為性連遺傳低磷酸佝僂症。在這種情形下如果雙親中母親是患者時,生育下有疾病的病童機率是50%,而且男女患病比率均等。如果雙親中父親是患者時,生育下男孩是健康的,不帶致病基因,但如果生育的是女孩,則一定會患病。但另外三分之一的患童則是因為偶發的基因突變所造成,也就是並無家族史,父母皆無此症狀。在此情況下,父母親再生下患兒的機率不高。但是無論是以上兩種情形造成的疾病。當病童在面臨結婚生子時,均必須請教遺傳專科醫師,以進行正確的遺傳諮詢及產前檢查。遺傳模式
治療原則
患者會因腎臟不斷的流失磷酸鹽,造成血液中的磷酸鹽濃度比一般人低,於是副甲狀腺素這類的荷爾蒙便會長期的刺激骨骼釋放出鈣離子及磷酸鹽,而造成骨質疏鬆及畸形,甚至嚴重者會經常性的骨折。性連遺傳低磷酸佝僂症的藥物治療已經行之多年,雖然治療效果因人而異差異性很大,但仍然傾向使用藥物治療。治療的目的可以促進生長速率、改善或是預防病童骨骼上的畸形、預防出現弓形骨以及減少骨頭的疼痛。不過在藥物治療方面,促進生長的效果較不如藥物對骨骼疾病方面的幫助較大。
主要藥物:高劑量的磷酸鹽、Carcitriol
磷酸鹽
藥物介紹:磷酸鹽類,以補充體內不斷流失的磷酸鹽。藥物有水溶液型及錠劑。可依據病童年齡給予適合的型態藥物。
Carcitriol
藥物介紹:1,25(OH)2-VitamineD3。
注意事項
1.藥物治療的開始時間,對於疾病的預後很重要。愈早治療其預後情況愈好,因此疾病的早期診斷相當重要。對於有家族史的病童,因及早檢查血液及尿液中是否有不正常的磷離子濃度,並且進行骨骼的X光攝影確認。最理想的診斷時間是出生後1個月至6個月之間。2.治療一般從嬰兒時期約2歲開始。開始治療時必須從低劑量開始給予磷酸鹽及Carcitriol,目的在於防止腹瀉。然後逐漸地提高藥物劑量,就是所謂高劑量期。典型的方法,當病童服用了一年的高劑量藥物後,會將藥物劑量調整為長時間持續服用的較低劑量的治療模式。
3.需依年齡選擇合適的藥物劑型。當病童年齡較小時,以考慮使用液態磷酸鹽藥物餵食,年齡大些可以調整成粉狀或錠劑藥物服用。
4.以上的藥物治療有時會伴隨一些副作用。服用高劑量磷酸鹽易造成的腎石灰沉著病及次發性副甲狀腺過高症。因此在高劑量期必須經常監控血中Ca2+、P3-及肌氨酸酐,同時每月要監測尿液中的Ca2+及肌氨酸酐。再過了高劑量期之後的長期服用期,則可以每3至4個月再檢查一次血液及尿液的數據。
5.目前藥物治療是否套用於成年人身上有很大的爭議。因為過了成長期的成人病患服用此藥物,雖然可改善生理症狀及組織結構,但效果不如兒童時期就進行治療來得有意義。
其它附屬的治療藥物:
1.利尿劑如Thiazidediuretics或amiloride,會被使用於治療性連遺傳低磷酸佝僂症的輔助藥物。主要目的在於增加鈣離子再吸收。
2.人類成長荷爾蒙
有報告顯示,人類成長荷爾蒙如果也成為標準的治療藥物,可以促進血液中循環的磷酸鹽濃度,因而幫助身材較矮小的病童再長高點,但效果並不顯著。
3.維生素D的類似物
可以控制副甲狀腺過高的副作用發生,用於輔助磷酸鹽藥物治療使骨骼生長方面達到最佳治療效果。
4.整型及骨科外科治療:
對於一些未定時服用藥物或來未能早期藥物治療的個案,當其骨骼產生嚴重變形或骨折時,須由整形外科治療,提供支架固定骨骼,或由骨科外科醫師手術介入治療。

