
簡介
血友病甲(hemophilia A,HA)是一種X染色體連鎖的凝血因子Ⅷ量和分子結構異常引起的隱性遺傳性出血性疾病,臨床特點為“自發性”關節出血和深部位組織出血。早在公元2世紀,猶太法典已有類似血友病遺傳的記載,男孩包皮環切術後出血,他的同母兄弟做包皮環切術有出血的危險。19世紀Schonlein開始使用血友病(hemophilia)這個名稱。
臨床表現
血友病甲的臨床特點是人體不同部位異常的過度出血尤其軟組織血腫和關節出血更是本病的特點出血的嚴重性與患者FⅧ:C的水平平行。血友病甲患者的出血與損傷有關,只是由於損傷極輕微而不被注意,而認為“自發性”出血。
我國制定的血友病分型標準,雖然在型之間存在某些重疊,但這種分型仍廣泛使用,在臨床診斷治療方面有重要意義。國際上分型標準並不一致。Hougie分為3型無亞臨床型,臨床特點與我國標準相似,但重型FⅧ:C小於1%,輕型FⅧ:C6%~30%。Wintrobe與我國標準除亞臨床型FⅧ:C25%~50%外其餘相同,還有幾種分型,差別健康搜尋在FⅧ:C水平的範圍有差別國外以Hougie分型使用較廣。重型血友病甲常經歷由日常活動引起的無明顯創傷的出血。兒童患者學步前無關節出血,以軟組織出血多見鶒開始走路後關節出血開始經常發生如果沒有有效的替代治療,反覆關節出血常可在患者成年前導致慢性血友病性關節病這是重型患者的特點。但是即便是重型血友病患者,其出血的發作也是間歇性的,數周數月甚至多年不發生嚴重出血並不少見。也有成年後才診斷的重型病例。除腦出血外,出血引起的突然死亡並不多見。中型病例可有血腫和關節出血,且常常由明確的創傷所引起。少數可有關節畸形,但很少在成年前出現。輕型病例極少有關節出血無關節畸形。
出血發作也不易發生,常有明顯的創傷引起許多患者僅有輕微鶒的易忽略的出血病史,經常因手術引起出血而得到診斷。多數攜帶者無出血症狀,FⅧ:C小於45%者可在手術和較大創傷後發生出血異常極少數FⅧ:C小於5%者臨床如同中型血友病甲,有時診斷為女性血友病。
1.關節出血是本病典型的出血症狀之一約見於2/3以上的病例。常發生在創傷行走和運動後。關節出血的好發部位以頻度排列依次為膝關節、踝關節肘關節、髖關節肩關節和腕關節。關節出血與該關節的承重和活動強度有關在學步前的兒童很少有關節出血。關節出血常來自該關節的滑膜血管,血液進入關節腔和骨骺等部位關節出血開始時有輕微的關節不適歷數分鐘到數小時,然後逐漸疼痛加劇關節局部腫脹發熱血液滲入皮膚或皮下時可有發紅和淤斑鶒關節活動受限。有時患者可有低熱鶒,但明顯持久發熱常提示合併感染。由於關節結構的關係,關節出血常呈自限性。當出血停止後,有些患者關節內血液健康搜尋在數天至數周內逐漸吸收關節功能逐漸恢復。反覆關節出血使關節造成慢性損傷滑膜炎關節軟骨破壞,骨質增生和萎縮,關節面唇樣增生和骨贅形成,關節腔狹窄以及骨質壞死和囊性變,導致關節畸形和功能障礙嚴重者造成跛行。若能對關節出血進行早期替代治療,常可使患者及早停止出血和疼痛並加快血液吸收,避免嚴重的血友病性關節病和患者致殘。血友病甲患者小關節出血發作很少而脊柱關節極少出血。
2.肌肉出血和血腫是血友病也是其他凝血因子缺乏症的特徵,其他出血性疾病少見。常在創傷或活動後發生,也可在創傷不明顯情況下發生。可發生在任何部位但用力肌肉群更易發生。約75%患者發生過肌肉出血和血腫。皮下和肌肉出血均有向四周擴散傾向,血腫可以逐漸增大,嚴重病例尤其是腹膜後出血可引起貧血和休克。血腫壓迫重要器官後果嚴重,腹膜後出血可以引起麻痹性腸梗阻,血腫進入胸腔或頸部可造成呼吸道阻塞,下腹部血腫導致尿路阻塞可影響腎功能血腫壓迫神經可致神經損害,髂窩部位的出血常是致殘的。肌肉出血的頻度依次為小腿、大腿、臀部前臂和腹部。
3.皮膚和黏膜出血皮膚和黏膜部位出血並非本病特點,其他出血性疾病也常有皮膚和黏膜部位的出血。血友病甲皮膚出血的特點是不表現為出血點,而呈片狀淤斑並常伴有皮下硬結,系真皮層以下部位出血形成的小血腫,常因輕微創傷引起。皮膚有較大傷口時常出血不止。黏膜出血常見,黏膜部位小傷口常引起持續地出血不進行替代治療不易停止齒齦、舌和其他口腔黏膜部位的小傷口常出血持續不止,若不進行替代治療,可以導致嚴重失血。消化道出血健康搜尋不少見,出血常嚴重,可因食物損傷上消化道黏膜或消化性潰瘍引起。成年血友病患者中消化性潰瘍發生率為正常男性的5倍。
4.假腫瘤發生率約為2%鶒,多見於重型血友病甲缺乏替代治療的患者。常見部位是大腿、骨盆和髂腰肌也可發生在臀部、小腿、足、前臂和手。局部創傷出血後在骨膜下、肌腱筋膜下形成囊性血腫若血腫內血液不吸收則血液破壞降解造成局部滲透壓增高,囊內反覆出血常在數年內體積逐漸增大,從而壓迫破壞和腐蝕周圍組織形成假腫瘤。假腫瘤是血友病危險的併發症,它可分3種類型。第1種為單純的囊腫,有蒂連於肌肉筋膜第2種開始時囊性,但由於影響附近骨和骨膜的血管供應,導致骨質吸收和囊腫形成。第3種是由於骨膜下出血導致骨膜和骨質的分離。
5.泌尿道出血重型血友病甲患者中常見。尿色可呈棕紅色或鮮紅色,由出血量多少而定出血部位一般在腎實質,多為單側,也可雙側腎同時出血下段尿路也可以發生出血。出血量一般不大。
6.手術後出血沒有進行替代治療的血友病患者手術常導致嚴重的出血。出血異常不僅在手術中,儘管已進行充分止血手術後數小時甚至數天后出現嚴重出血很常見手術切口常不癒合或癒合不良。無論是大手術或小手術,必須在術前就開始替代治療直至傷口癒合。拔牙後出血很常見,但是乳牙自然脫落很少引起過量出血潔齒和其他牙科操作在沒有替代治療的患者也可引起嚴重的出血。6-氨基己酸類纖溶抑制劑可減少FⅧ製劑的用量肌內注射可引起注射部位巨大血腫。各種因創傷引起的傷口縫合後出血常見常需替代治療才能止血 併發症: 顱內出血及周圍神經系統症狀是本病最常見的併發症,顱內出血也是最常見的致死原因。多有外傷史,但有時外傷輕而沒引起注意出血部位可在硬膜外、硬膜下及腦內。可表現為逐漸加重的頭疼鶒,逐漸發生昏迷以及顱內壓增高的症狀和定位體徵。許多患者在外傷後數天才出現中樞神經系統症狀因此對有頭部外傷可能腦出血的患者應及早替代治療。腦電圖異常提示以前可能發生過亞臨床健康搜尋的腦出血。周圍神經系統常因出血侵及或血腫壓迫導致劇烈疼痛、麻木和功能障礙,肌肉萎縮。
治療
血友病甲患者在進行血友病的治療時,還可能涉及骨科,普通外科以及口腔科等方面,而當血友病患者患有其他疾病需要治療時也應當考慮血友病可能產生鶒的影響。阿司匹林,非腎上腺皮質激素類抗炎藥物以及影響血小板聚集功能的其他藥物,應儘可能不用於血友病患者。肌內注射通常應當禁止血友病患者發生出血症狀時治療越早越好可以避免出血併發症的出現,不僅比延遲的治療效果好,而且可減少替代治療次數。家庭成員應儘可能了解血友病,家中應備有替代治療藥物以便隨時可用在有條件時進行家庭治療和預防治療,並得到血友病診斷治療中心的幫助迄今血友病甲最有效的治療方法是替代治療,即輸注FⅧ製劑其他藥物包括止血藥和一些療效不肯定的藥物如花生衣等對血友病的出血沒有多大作用,不能因為使用了這些藥物而不用替代治療從而導致出血延續,並引起併發症的發生。血友病患者肌肉強壯可以減少或減輕出血的發作,因而應在醫生指導下適當進行肌肉鍛鍊。
1.FⅧ替代治療替代治療是血友病甲出血發作時健康搜尋的主要治療。由於FⅧ製劑的大量生產和普遍使用血友病患者的平均壽命已接近正常人。一些已開發國家已對兒童進行每周1次的預防治療,成年後進行出血時治療使該病的關節病和致殘率大大降低。FⅧ製劑中FⅧ含量用國際單位(IU)或單位(U)度量。1U定義為用3.8%枸櫞酸鈉1∶9抗凝的1ml平均正常人血漿所含FⅧ所具有的凝血活性用國際標準品標定的參照標準品所測出的單位(U)稱為國際單位(IU)。臨床上國內外多習慣於用百分比表達FⅧ:C的水平,100%=IU。FⅧ體內半衰期10~12h,應每12小時輸注1次。替代治療遵循早治,足量和維持足夠時間的原則進行。一般認為每公斤體重輸注IUFⅧ能提高FⅧ:C水平2%,每次需要輸注的劑量可按下述公式計算:需FⅧ量(U)=(期望FⅧ:C-患者FⅧ:C)×體重(kg)/2.如1個50kg體重患者FⅧ:C小於1%,期望FⅧ:C提高20%,則需要輸注FⅧ=(20-0)×50/2=500U。出血的嚴重性和部位健康搜尋不同所需輸注量也有差別。由於FⅧ製劑價格昂貴多數醫生喜歡用能達到有效止血的最小量。表3列出了一些常見出血的FⅧ製劑的期望值需要輸注量以及治療的持續時間。目前國外使用的替代治療商品製劑品名很多。國內也已有病毒滅活或去除病毒製劑的生產表4列出了替代治療製劑的類型和一些特點,包括其他遺傳性凝血因子缺乏症的治療製劑。新鮮全血目前已很少用於血友病甲治療,除可以傳染血液傳播性病毒外提高FⅧ水平有限,常不能達到有效止血。新鮮血漿也由於同樣的原因很少用於血友病甲的治療,但對沒有提純製劑的FV,FⅪ和FⅫ缺乏症仍是主要的治療方法。冷沉澱能達到止血要求,但不易病毒滅活,每袋含量不穩定健康搜尋,需要冰凍保存血液製品病毒傳染是嚴重的問題,我國已規定禁止無病毒滅活和去除病毒工藝鶒的血製品生產,國內供應的FⅧ製劑和凝血酶原複合物均已去除病毒,使用是安全的。病毒滅活和去除病毒可用冷凍乾燥後80℃加熱,有機溶劑-去污劑滅活或用單克隆抗體提純因子後再經上述工藝滅活病毒處理後的血液製品目前認為不傳染病毒性疾病。重組DNA技術生產的FⅧ已有商品供應,安全有效,但價格昂貴。豬FⅧ由於與人抗FⅧ抗體無交叉反應適用於抗Ⅷ抗體的血友病甲患者的治療。
2.1-去氨基-8-D精氨酸加壓素(DDAVP)這是一種部分合成的加壓素衍生物,有抗利尿和使內皮細胞釋放vWF和FⅧ等的作用,可使正常人及輕中型血友病甲患者FⅧ:C暫時性升高。大多數病例第1次用DDAVP0.3μg/kg可使FⅧ:C升高2~3倍,高峰在給藥後30~60min內出現。重型血友病甲患者對DDAVP無反應一般每12小時給藥1次。每次給藥釋放FⅧ量逐漸下降,一般第3天后FⅧ上升不明顯應停藥DDAVP可作為輕型和一些中型血友病患者在某些不需要較高FⅧ:C情況下取代替代治療的用藥。副作用有心率加快,顏面潮紅,水瀦留少見但仍應預防攝入過量的液體。
3.其他藥物治療(1)纖溶抑制藥:如氨基己酸有輔助血友病止血的作用可使已形成的少量凝血塊不被纖溶作用所溶解。在黏膜出血特別是拔牙後出血較有價值,可以減少FⅧ製劑的用量氨基己酸成人可用4~6g,3次/d但必須指出泌尿道出血患者禁用纖溶抑制類藥物,以避免造成泌尿道阻塞。(2)達那唑:是一種部分合成的雄性激素,也曾試用於血友病甲和血友病乙,目前認為僅對極少數患者有效,特別是血友病乙基因突變發生在啟動子部位健康搜尋的變異型Leiden,雄性激素有治療作用。(3)腎上腺皮質激素:可減輕出血引起的局部炎症反應鶒,加速血液吸收,有時也可用於血友病患者。對泌尿道出血皮質激素療效較好有些患者單用皮質激素和臥床休息可使出血停止。常用地塞米松(氟美松)5~10mg/d靜脈給藥。也可用潑尼松40mg/d由於皮質激素的副作用,療程不宜長。
4.局部止血治療輕微皮膚傷口出血和鼻出血可試用吸收性明膠海綿、止血纖維或棉球、局部止血藥凝血酶等壓迫止血。若壓迫止血無效則需替代治療。大的傷口出血或黏膜小傷口出血壓迫止血往往無效,需替代治療。
5.關節出血目前主張儘早替代治療腎上腺皮質激素有利於血腫吸收。關節積血沖洗有爭議健康搜尋,有的認為並無益處健康搜尋,有的認為可加速積血清除對預防和減緩關節損害有益。但在無充分替代治療情況下進行關節穿刺和沖洗肯定有害無益,將加重關節出血並可能誘發感染。
6.手術血友病患者在充分替代治療的條件下理論上可進行各種正常人所作的手術治療其他疾病。血友病的關節併發症也可進行滑膜切除術或關節置換術治療,但應掌握適應證,並考慮關節置換術後長期進行預防性替代治療的能力以及權衡手術後取得的療效是否優於手術前。
7.肝移植已有肝移植治療血友病甲病例的報導,以前認為僅試用於肝病晚期的病例,由於肝臟提供者難以解決,這種治療方法極少採用。
8.基因治療最近分子生物學研究的進展使基因插入治療的可能性增強這是血友病甲最為理想的治療前景但目前所作的研究仍處於初期階段作為一種臨床常規使用的治療手段仍需大量的工作。
9.替代治療是主要治療血友病甲的方法但也可引起嚴重後果。
(1)肝炎:1985年以前已開發國家幾乎所有血友病患者感染1種或1種以上病毒性肝炎病原體。其中,至少50%患者發展成為慢性持續性或慢性活動性肝炎,導致肝硬化。C型肝炎和B型肝炎最常見。自採用病毒滅活的替代治療製劑後(1985),肝炎傳染已大大減少,目前認為這種製劑是安全的。α-干擾素對C型肝炎和B型肝炎治療有一定的療效,但遠期療效並不明確。
(2)獲得性免疫缺陷病(AIDS愛滋病):1985年以前已開發國家中反覆輸注FⅧ製劑健康搜尋的血友病患者中約90%存在人類免疫缺陷病毒(HIV)抗體曾感染過HIV。感染HIV成了已開發國家中血友病患者預後健康搜尋的決定因素。但從1985年採用病毒滅活技術以來,替代治療製劑傳播AIDS的危險性已經消除。
(3)抗FⅧ抗體:反覆替代治療的血友病患者中,有3.5%~20%患者產生抗FⅧ的同種抗體鶒抗體有兩種類型一種為低反應型抗體,抗體滴度在反覆輸注FⅧ製劑後也不超過10Bethesda單位另一種為高反應型抗體,對FⅧ的再次輸注有回憶反應,抗體滴度上升到10Bethesda單位以上典型回憶反應是在再次輸注FⅧ後3~8天,抗體滴度開始上升,8~15天達到高峰。替代治療過的血友病患者應檢查抗體篩選試驗,在手術治療前則必須檢查。抗體引起的急性出血的治療需用極大劑量的FⅧ或豬FⅧ,或激活的凝血酶原複合物(APCC),常幾種製劑同時使用大劑量FⅧ和APCC常規治療可達到免疫耐受,但需要數月的不間斷治療,費用昂貴。
流行病學
 血友病甲
血友病甲據世界衛生組織(WHO)和世界血友病聯盟(WFH)1990年的聯合會議報導,全球血友病的發病率為15/10萬~20/10萬,其中血友病A占85%,血友病B占15%。血友病A的發病沒有地域與種族間的差異 歐美中國血友病的發病率平均為5/10萬~10/10萬,其中,美國為10/10萬,英國為6.9/10萬,瑞典為6.6/10萬,非洲為5/10萬,法國為6.3/10萬(馬提尼克島為16.1/10萬);血友病A法國占77.4%,土耳其占95%,澳大利亞占64.3%。中國缺乏對血友病的全面統計。據邵宗鴻等的綜合報導,血友病的發病率為2.27/10萬~2.84/10萬,其中血友病甲為1.95/10萬~2.40/10萬,約占85%。隨著篩選試驗和檢測方法的迅速發展,對輕型血友病患者,尤其是致病基因攜帶者的發現增多,發病率有升高的趨勢。
女性血友病甲患者極其罕見,雖然已有患病父親和攜帶者母親女兒患有血友病甲的報導,但所報導的女性患者中有相當部分是攜帶者女性,由於其正常X染色體極端隨機滅活導致有血友病的臨床表現;另外也可能是2N型血管性血友病。
與血友病患者有血緣關係的女性攜帶血友病基因的可能性分為3種:①肯定攜帶者,包括血友病患者的女兒、至少2個以上血友病兒子的母親;有1個血友病兒子的母親另外與她有母系血緣關係的女性中有另1個血友病患者;②很可能攜帶者,無遺傳家族史的血友病患者成為散發病例,散發病例的母親為很可能攜帶者;③可能攜帶者,與血友病患者有母系血緣關係且沒有血友病兒子的女性為可能攜帶者。
發病機制
FⅧ基因的缺陷導致FⅧ合成的障礙以及FⅧ分子結構的異常引起FⅧ功能活性的降低或缺乏是血友病的根本病理生理基礎。FⅧ基因和血友病甲的基因缺陷,FⅧ基因位於X染色體長臂末端(Xq28)包含186kb,約占X染色體全長的0.1%。外顯子26個,總長度約9kb。內含子25個。mRNA長約9029bp,,引起血友病甲的FⅧ基因缺陷可以是點突變、基因的部分或全部缺失、基因成分插入、產生終止密碼的點突變和基因倒位Tuddenham等總結的資料中各種不同的點突變超過80種,插入6種,小的缺失7種,大片段缺失60種。
目前已有近400種FⅧ基因缺陷導致血友病甲。在點突變中健康搜尋發生在限切酶位點單一鹼基突變數量多,Taq1識別TCGA序列,這一位點的突變可以通過1個Taq1斷裂位點的丟失直接得到測定。突變也常發生在CpG二核苷酸序列。精氨酸的密碼CGA常受這一序列突變的影響。CG二核苷酸序列發生C→T轉換,則CGA突變為TGA這是終止密碼,自此以後的蛋白質不再合成常常導致重型血友病。產生終止密碼的突變稱為無義突變。CG二核苷酸序列發生G→A轉換則CGA突變為CAA,這是谷氨酸的密碼健康搜尋,精氨酸被谷氨酸替代導致不能正常激活的無功能FⅧ分子,其FⅧ:Ag正常而FⅧ:C小於1%。
產生胺基酸被別的胺基酸取代的突變成為錯義突變。已有許多錯義突變報導,不同錯義突變所引起血友病的嚴重程度有差異。約5%的血友病甲由基因缺失引起並常導致重型血友病已發現的基因缺失分布在整個FⅧ基因,沒有特別的區段更傾向於發生缺失突變缺失長度可自2bp~210kb以上小的缺失若不改變基因的閱讀框架可導致輕型血友病。大的缺失常導致患者測不到FⅧ:Ag。雖然其他基因缺陷的患者也發生抗FⅧ抗體,但無FⅧ:Ag的大片段基因缺失的患者更易產生抗FⅧ抗體。插入是FⅧ基因缺陷中另一種突變類型。部分人類L1序列插入FⅧ基因可引起血友病健康搜尋。L1序列是一廣泛存在於人類基因組中的較長的重複序列,與反轉錄病毒反轉錄酶的DNA相似,由於能在基因組中移動又稱為轉座子(transposon)。在FVⅢ基因內插入轉座子導致血友病甲,已有一些病例的報導。基因倒位健康搜尋是近幾年發現的又一類血友病甲的基因改變。
FⅧ基因內含子22結構中有1個CpG島(雙向轉錄活性啟動子),與此島相關的有2個FⅧ基因的相關基因其1為F8A基因,其轉錄方向與FⅧ基因轉錄方向相反,另一個為F8B基因,其轉錄方向與FⅧ基因轉錄方向相同F8A基因在X染色體末端(Xq)有三個拷貝,1個位於FⅧ基因內含子22內(基因內基因),另2個位於FⅧ基因上游500kb處健康搜尋FⅧ基因內鶒的F8A可與上游2個F8A基因中的任1個發生染色體內的同源重組。重組使Xq末端發生倒位,將FⅧ基因分為方向相反的兩部分啟動子至外顯子22為一部分,外顯子23至外顯子26為另一部分,兩者相距約500kb,從而導致重型血友病甲。現在認為FⅧ基因倒位約占重型血友病甲的50%
血友病診斷
 血友病甲
血友病甲1.診斷標準(首屆中華血液學會全國血栓與止血學術會議修訂,1986)
(1)臨床表現:①男性患者,有或無家族史。有家族史者符合性聯隱性遺傳規律 女性純合子型可發生,極少見;②關節、肌肉、深部組織出血,可自發。一般有行走過久、活動用力過強、手術(包括拔牙等小手術)史。關節反覆出血引起關節畸形,深部組織反覆出血引起假腫瘤(血囊腫)。
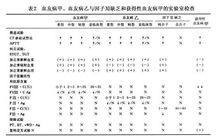
(2)實驗室檢查:①凝血時間(試管法)重型延長,中型可正常,輕型、亞臨床型正常;②活化部分凝血活酶時間(APTT),重型明顯延長,能被正常新鮮和吸附血漿糾正,輕型稍延長或正常,亞臨床型正常;③血小板計數 出血時間、血塊回縮正常;④凝血酶原時間(PT)正常;⑤Ⅷ促凝活性(FⅧ:C)減少或極少;⑥血管性血友病因子抗原(vWF:Ag)正常 FⅧ:C/vWF:Ag明顯降低
(3)嚴重程度分型(表1)。
(4)排除FⅧ抗體所致獲得性血友病甲(獲得性FⅧ缺乏)。
2.產前和攜帶者檢查 對可能是攜帶者的家系成員進行基因檢查在遺傳諮詢時具有重要意義,但是,必須指出約有30%患者的突變是自身新發的而非遺傳所得。重組DNA技術為產前診斷和攜帶狀態的檢查提供非常有利的手段 可進行限制性片段長度多態性(RFLP)分析的家系必須包括1個男性患者,並且其母在1個RFLP標誌上是雜合的。
(1)產前診斷:①FⅧ:C和vWF測定:以往主要依靠妊娠18~21周時,通過胎兒鏡取胎兒血檢測其FⅧ:C和vWF水平,可是1978~1983年,在美國套用這種方法進行產前診斷只有92例,而每年估計有300個胎兒有血友病甲的危險性。這種產前診斷方法之所以不被廣泛採用的原因是胎兒鏡檢查引起流產者可達6%。採血失敗率可達13%(12/92)。②FⅧ的RFLP:提供一種產前診斷的可靠方法。有2種提取胚胎DNA的方法:對3月齡胚胎進行穿刺術取樣;對8~11周的絨毛膜取樣。要滿足RFLP的條件,即必須有先證者。先證者的母親必須是該酶切位點的雜合子。用PCR技術使RFLP分析大大簡化 但是,有某些局限,有的家族無DNA標誌可用,只能單靠FⅧ:C和vWF的檢測。
(2)攜帶者檢查:男性患者與正常女性所生兒子均為正常,所生女兒均為攜帶者;女性攜帶者與正常男性所生的兒子有50%機率為血友病患者,所生的女兒有50%機率成為致病基因攜帶者;女性攜帶者與男性患者所生的兒子有50%機率為血友病患者,所生的女兒要么是致病基因攜帶者,否則就是血友病患者;男性患者與女性患者所生的兒子和女兒都是患者,幸運的是這種機率極為罕見。
鑑別診斷
1.經典的血友病甲需要與血管性血友病(vonWillebrand’sdisease,vWD)相鑑別vWD的發生與FVⅢ在體內的載體vonWillebrand因子(vWF)缺乏有關。因此,在vWD中FⅧ的水平也下降,雖然下降的幅度在不同的患者中可能有較大健康搜尋的差異。在vWD的患者中,FVⅢ健康搜尋的合成雖然是正常的,但是,由於它的載體vWF水平下降而在體內的半衰期縮短。將vWD與血友病甲鑑別開來的其他表現有出血時間延長、vWF抗原水平下降、瑞斯托黴素誘導的血小板聚集下降等。
2.與其他造成APTT延長的遺傳性凝血因子缺乏性疾病相鑑別如凝血因子Ⅸ、Ⅺ、Ⅻ,激肽釋放酶原,高分子量激肽原的缺乏等。只有因子Ⅷ缺乏和因子Ⅸ缺乏的病例表現有X連鎖遺傳的特點,並且也只有這兩種因子缺乏會累及關節,造成殘疾。血友病甲只有通過特異的檢查才能與血友病乙鑑別開來。男性或女性都可發生因子Ⅺ缺乏而且與經典的血友病相比,出血較輕。
3.由於FⅫ、激肽釋放酶原高分子量激肽原的缺乏不會造成臨床出血的表現,因此,很容易與經典的血友病相鑑別。輕型的血友病(FⅧ水平大約為正常的15%)必須考慮到有因子Ⅴ與因子Ⅷ聯合缺乏的情況,雖然APTT和PT都可能有輕微的延長但是除非考慮到這種聯合缺乏,否則常常得不到正確的診斷。
 血友病甲
血友病甲4.獲得性因子Ⅷ缺乏(獲得性血友病甲)多由於血液中有抗FⅧ抗體存在所致,其出血的臨床表現與血友病甲基本相同但是,出血程度往往較重。本病可發生於以往健康者、女性(尤其妊娠期)、老年人,以及某些免疫性疾病患者。實驗室檢查方面,APTT和S-CT延長,且等量正常血漿不能糾正STGT或BTGT的缺乏,檢測抑制物(抗FⅧ抗體)滴度增高對鑑別更為準確臨床常用Bethesda方法檢測健康搜尋血友病甲、血友病乙與因子Ⅺ缺乏和獲得性血友病甲的實驗室檢查的特點見。
5.血友病甲導致的出血和血腫等症狀在診斷未明確時還可能與其他一些疾病相混淆如將深部血腫誤認為化膿性病灶而施行切開引流;髂腰肌出血誤為闌尾炎;將腹膜後血腫誤診為闌尾膿腫;將血友病甲關節出血誤為結核、關節炎和肉瘤等將血友病引起的出血或血腫誤為腎腫瘤、肺部疾患、消化道潰瘍,腹腔內出血當作潰瘍穿孔腸梗阻等均有報導。
