病因概述
 小腦性共濟失調
小腦性共濟失調此後在亞速爾群島、葡萄牙本土、巴西、美國、加拿大、日本等地先後發現很多葡萄牙起源的MJD家系。葡萄牙本土的MJD患病率為0.6/10萬人口,其中亞速爾群島為40.8/10萬人口,Flores島更是高達9/1000。最初的研究認為MJD僅限於葡萄牙及葡萄牙後裔,隨著更多家系在世界範圍內被發現,目前已證實MJD在五大洲內均有分布,而且很多家系並不具有任何葡萄牙血緣關係。在葡萄牙起源的SCA患者中,SCA-3/MJD陽性率高達41%~74%,為發病率最高的常染色體顯性遺傳性共濟失調。國內外研究發現即使是在非葡萄牙血統的人群中,SCA-3/MJD陽性率也高達17%~55%,同樣為SCA最常見基因型;而SCA-1、SCA-2、SCA-6、DRPLA的陽性率分別為SCA的3%~27%、5%~13%、6%~27%及1%~20%,這些研究結果的差異與種族差異和遺傳背景不同有關。目前較一致的觀點認為:SCA-3/MJD是最常見的一種SCA亞型,而DRPLA主要見於日本人群,SCA-7主要見於美國、法國等人群。
發病機制
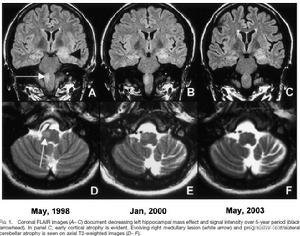 馬查多-約瑟夫病
馬查多-約瑟夫病MJD-1基因由1776個鹼基(bp)組成,其間含有一個較長的開放編碼結構,CAG重複位於開放編碼區的C端,並編碼產生一段多聚谷氨醯胺序列。在CAG重複序列內部有三處可被兩種可變序列(CAA或AAG)打斷。Northern印跡顯示所有組織中均有1776bpmRNA微量表達,但睪丸中有很強的2kbmRNA表達;而逆轉錄PCR(RT-PCR)顯示人腦中這兩種不同CAG重複長度的mRNA均有表達;這些結果提示兩種等位基因均在人腦中表達,而且每個等位基因的CAG重複次數都具有多態性。但MJD患者不同腦組織層面的MJDl基因mRNA水平並沒有顯著性差異,說明選擇性部分腦組織神經元變性存在其他調節因素。
MJDl基因的編碼產物ataxin-3蛋白為一種含有多聚谷氨醯胺(polyglutamine,polyGln)的胞漿蛋白,正常功能尚不明確。當ataxin-3蛋白含有異常擴展polyGln肽鏈後,可能獲得新的功能,引起神經系統內特定區域的神經細胞核內包涵體(neuronalintranuclearinclusions,NIIs)形成和神經細胞變性脫失。與其他由CAG三核苷酸重複動態突變引起的神經系統變性病如SCAl、亨廷頓舞蹈病(HD)、DRPLA研究類似,含有異常擴增(CAG)n的疾病基因編碼產生異常擴增polyGln肽鏈的蛋白後,在胞漿中被半胱天冬酶(caspase)等水解形成不同分子量大小的含異常擴增polyGln殘基片段,此類片段進入細胞核內,並不斷聚集形成核內包涵體,同時導致細胞死亡。研究發現只有被截斷的含異常擴增polyGln殘基的ataxin-3cDNA片段方可進入核內,聚集形成核內包涵體和引起細胞死亡;而含正常polyGln或含異常擴增polyGln的全長ataxin-3cDNA則沒有此類改變。這一結果在轉染細胞和轉基因動物的研究中均已得到證實。
推測這種選擇性神經系統損害可能與含異常擴增polyGln的突變蛋白在胞漿中與某特異性蛋白發生蛋白-蛋白相互作用,並最終導致選擇性神經元細胞NIIs形成和變性脫失有關。這種相互作用可能與polyGln兩側的功能域(domain)序列有關。
基因結構
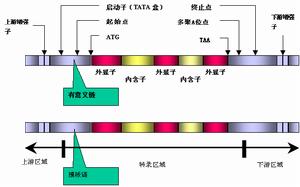 MJD-1基因結構圖
MJD-1基因結構圖對不同地域及人種的SCA-3/MJD家系研究已經發現:
①異常擴增的CAG重複數大小與患者發病年齡和疾病嚴重程度呈負相關,即(CAG)n重複擴增數越大,發病年齡越早,臨床症狀越嚴重。
②CAG重複數存在代間不穩定性,重複數增加的情況多於減少。這為SCA-3/MJD患者臨床上的遺傳早現現象提供了合理的解釋,即子代發病提早和病情加重是由於三核苷酸重複拷貝數在傳代過程中增加的結果。同時,大部分研究發現SCA-3/MJD存在父系遺傳傾向,即父系遺傳時CAG重複擴增數目增加的幅度較大,但這種不穩定性遠不如HD、SCA-1和DRPLA那么明顯。進一步研究SCA-3/MJD男性患者精子中擴展的CAG重複數目時,發現有92%與同一患者外周血自細胞中的CAG重複數不同,其中32%表現為增大、60%為縮短,說明代間不穩定性可能也與男性患者減數分裂中擴增的等位基因分離不平衡有關。
③CAG重複數與某些臨床症狀、體徵的出現存在相關性。如在Takiyama的研究中,CAG擴增重複數目與突眼和錐體束征呈正相關,而Durr則發現腱反射異常、振動覺消失和軸突型神經病與CAG擴增重複數存在一定相關性,但並不足以解釋個體間的症狀差異。由於主要臨床症狀、體徵與CAG重複數目相關性的研究在不同家系得到的結果並不一致,而且CAG重複數只能是疾病的遺傳標誌,影響疾病的表型還存在其他修飾因素,因而不能完全將CAG重複數作為SCA-3/MJD臨床表現的預測指標。
病理
 脊髓小腦束
脊髓小腦束與SCA-1比較,SCA-3/MJD患者基底節損害更為嚴重,特別是內側蒼白球和下丘腦核的變性更為明顯,而SCA-1蒼白球變性以外側為主。相反,橄欖和小腦皮質損害主要見於SCA-1,表現為蒲肯野細胞脫失和小腦下腳變性。另外,儘管兩者都有脊髓損害,部位卻不盡相同:後柱受累多見於SCA-1,Clarke柱受累多見於SCA-3/MJD,而中間外側柱變性則只在SCA-3/MJD存在。與DRPLA相比,後者通常沒有眼球運動神經核細胞脫失,蒼白球變性也僅限於外側。
值得注意的是,雖然SCA-3/MJD的病理改變具有一定的特徵性,但在不同病例各部位受累程度可能不同,而且有時也存在異質性。如兩例中國SCA-3/MJD患者病理改變上,蒼白球、路易氏體和齒狀核受累較輕,黑質、紅核、腦神經運動核、Clarke柱、脊髓小腦束、脊髓前角細胞受累較重。另外,Cancer報導的病理改變符合DRPLA的病例,基因診斷卻為SCA-3/MJD。
臨床表現
 突眼
突眼SCA-3/MJD患者主要臨床表現有小腦性共濟失調、構音障礙、吞咽困難反嗆、痙攣狀態、錐體束征等,可伴有眼球突出、面舌肌搐顫、突眼、注視麻痹、慢眼活動、肌萎縮、肌張力障礙、錐體外系體徵、自主神經症狀等。
Coutinho(1978)將SCA-3/MJD分為三型:I型以肌張力障礙—強直性錐體外系症狀、錐體束征和進行性眼外肌麻痹為主,Ⅱ型以小腦體徵和錐體束征為主,Ⅲ型以遠端對稱性肌萎縮和小腦體徵為主,周圍神經病體徵明顯,包括肌無力、肌萎縮和感覺遲鈍等。次要症狀如進行性眼外肌麻痹、面舌肌搐顫和突眼雖然不常見,但都是SCA-3/MJD的特徵性表現。各型患者發病年齡分別為:I型平均24.3歲,Ⅱ型平均40.5歲,Ⅲ型平均46.8歲;CAG擴展重複數分別為:I型79.4±1.O,Ⅱ型74.6土O.5,Ⅲ型72.6±1.1;CAG重複數與發病年齡呈明顯負相關。Rosenberg補充的第Ⅳ型為老年發病、明顯的帕金森征象伴共濟失調、遠端肌萎縮和感覺消失,部分Ⅳ型患者開始被診斷為帕金森病。另外,SCA-3/MJD也可以痙攣性截癱的表型出現,一些臨床表現為典型痙攣性截癱的患者經基因診斷證實為SCA-3/MJD。當然,很多病例並不是以某一亞型獨立存在,而是表現出明顯的型間重疊和過渡,即在病程不同階段,表現出不同亞型的臨床症狀。
診斷
 第二節血細胞
第二節血細胞在第3屆國際MJD會議上,Nicholson報導了一個澳大利亞家系,臨床表現為MJD,但有SCA-1基因突變;Larrarini則發現一個定位於染色體14q32.1的法國大家系,三代分別出現MJD、SCA-1和脊髓腦橋型共濟失調的臨床症狀;Cancel同樣在法國發現一個表型為DRPLA的家系存在MJDl基因突變。而經基因檢測確診的中國SCA-3/MJD患者中,少數出現智慧型障礙和腱反射減弱,很難在臨床上與SCA-2及部分SCA-1患者區分。因此,雖然SCA-3/MJD具有一定的臨床特徵,但由於其明顯的臨床異質性,僅僅根據臨床表現很難作出正確的診斷,只能藉助於基因診斷確診。
輔助檢查
 小腦蚓部
小腦蚓部SPECT顯示小腦局部腦血流(rCBF)明顯減少,但rCBF降低程度與MRI顯示的小腦萎縮程度不一致。PET檢測除小腦半球、小腦蚓部和腦幹外,枕部皮質也有明顯的局限性低代謝。
SCA-3/MJD患者的體感誘發電位和聽覺誘發電位異常。經顱磁刺激研究發現SCA-3/MJD患者的運動誘發電位幅度明顯異常,SCA-1患者的中央運動傳導時間延長、運動誘發電位閾值提高,而SCA-2患者則很少異常。提示電生理檢查在SCA的臨床鑑別中有一定意義。
另外,通過眼球運動檢測(快速掃視眼動振幅、快速掃視眼動速率、凝視誘發眼震的存在與否)也可從臨床上初步鑑別SCA-1、SCA-2及SCA-3/MJD:其中SCA-3/MJD患者存在凝視誘發眼震,SCA-1患者的掃視振幅明顯增高,而SCA-2患者的掃視速率顯著下降。
基因診斷
由於MJDl基因(CAG)n重複數在正常人和患者之間存在明顯差異而且沒有重疊,因此選用合適的引物進行聚合酶鏈式反應(PCR)擴增,可作出準確的基因診斷。通過PCR擴增技術獲得含有MJDl基因(CAG)n結構在內的PCR擴增片段,如果其中有或至少有一條PCR產物片段大小超出了正常範圍,則可診斷為SCA-3/MJD患者或症狀前患者。根據文獻報導結果,正常人MJDl基因兩條等位基因的CAG重複數均在12~41次之間,而患者至少有一條等位基因的CAG重複數在56次以上,大部分在62~84次之間。但確切的CAG重複數必須通過測序分析獲知。
