病因
 肛門直腸先天畸形
肛門直腸先天畸形肛門和直腸的發育,發生在第4周~6個月期間,胚胎長度為4~200mm階段。在胚胎4mm時,後腸擴大與尿囊相通,形成泄殖腔,為一盲囊,中腎管開口於泄殖腔內。胚胎5mm時,泄殖腔與尾腸延伸相通。在體壁的腹側有泄殖腔膜,系外胚層與內胚層相融合的很薄組織,使泄殖腔和體外相隔。第4周泄殖腔開始分成兩部分,背側部形成直腸,腹側部稱為尿生殖竇。其分隔過程是中胚層組織向尾側方向生長,稱為Tourneaux′s褶。間質從側壁向內側方向增生,稱為Rothke′s襞。兩種組織結構在中間融合形成尿直腸膈。
隨著泄殖腔的分隔,泄殖腔膜也被分為前後兩部分,前面為尿生殖竇膜,後側成為直腸膜,並構成原始會陰。在第7周末,尿生殖竇向外開口,第8周時直腸肛膜破裂。在此之前,從第5周開始,外胚層向內發展形成肛凹,並逐漸加深接近直腸,最後兩者相通。
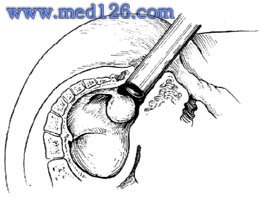 肛門直腸先天畸形
肛門直腸先天畸形生殖器官和會陰的形成與上述發育過程同時進行。在女性:內生殖器官是由苗勒管形成,向下延伸至中胚層的泌尿生殖隔深處,其中段和下段融合,形成子宮和陰道,未融合部分形成輸卵管,中腎管退化。生殖襞的後半部與尿直腸隔的會陰距狀突融合,形成會陰和叉狀的陰道前庭原基,其前半部形成小陰唇,生殖隆凸未融合而形成大陰唇。在男性:內外生殖器和會陰形成時,睪丸發育,中腎管形成輸精管,苗勒管退化。生殖襞向中線靠近覆蓋尿生殖竇孔,並逐漸癒合形成球部和陰莖部尿道。生殖隆凸形成陰囊。在胚胎第4個月時,會陰向前後方向迅速增長,使肛門移到正常位置。
直腸肛門畸形的發生是胚胎發育期發生障礙的結果,男性和女性基本上是相同的,僅是解剖上的區別。泄殖腔分隔過程的結果,尿生殖竇與肛門直腸竇之間相通,構成高位或中間位畸形,發生各種肛門直腸發育不全及直腸與尿道或陰道間的瘺管。肛門後移過程障礙和會陰發育不全的結果,構成低位畸形,發生肛門皮膚瘺,肛門前庭瘺,肛門狹窄等。
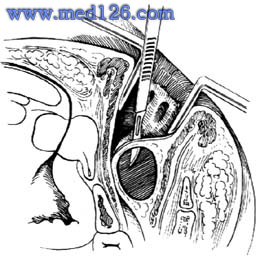 肛門直腸先天畸形
肛門直腸先天畸形肛門直腸的局部解剖和排便控制機理是個複雜問題。新生兒肛管長度1.2cm(0.8~2cm),直腸長度5.2cm(3.5~7.5cm),近肛門處存在前凸的直腸會陰曲,肛管縱軸與會陰平面交角85°(60~90°)。腹膜反折距肛門約2.9cm(2~4cm),骨盆神經從位於骶前距肛門約3cm。直腸末端的環肌束向下延伸並增厚形成的括約肌,是平滑肌,呈不自主的收縮狀態,以閉合肛門。環繞直腸的外括約肌是橫紋肌,可隨意志而收縮,包括尖頂袢、中間袢和基底袢三個肌群。肛外括約肌與恥骨直腸肌應視為一個統一體,共同構成控制排便的三個重要肌環,稱為三環系統(tripleloopsystem)。具有直接反向壓迫作用和絞鎖機制,對肛門進行有力的控制。三環袢完全損傷,即導致肛門失禁。在肛管盲腸畸形時,各肌纖維發育不正常,纖維走向亦有改變。外括約肌包括恥骨直腸肌肌纖維稱為橫紋肌複合體(striatedmuscularcomplex),以利手術進行。直腸恥骨肌在直腸肛管交接處形成一個環狀吊帶,稱為恥骨直腸環(Pubo-rectlsling),收縮時增加直腸下段的壓力,起到排便的控制作用。盆腔肌左右兩束肌群總稱肛提肌,形成小骨盆腔內協調的收縮作用,幫助排便。聯合縱肌是內、外括約肌間的纖維性組織,起固定肛管作用並協助排便。肛管直腸畸形手術時,應儘量保持肛門直腸周圍肌群的完整性,以獲得較完善的排便功能。
病理
 肛門直腸先天畸形
肛門直腸先天畸形臨床表現
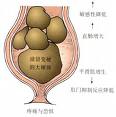 肛門直腸先天畸形
肛門直腸先天畸形(一)高位畸形約占40%,男孩多見,往往有瘺管存在,但因瘺管細小,幾乎都有腸梗阻症狀。骨盆肌肉的神經支配常有缺陷,並伴有骶椎和上尿路畸形。此型病例在正常肛門位置皮膚稍凹陷,色素較深,但無肛門。哭吵時凹陷處不向外膨出,用手指觸摸也無衝擊感。女孩往往伴有陰道瘺,開口於陰道後壁彎窿部。外生殖器亦發育不良,呈幼稚型,糞便經常從瘺口流出,易引起生殖道感染。男孩常伴有泌尿系瘺,從尿道口排出氣體和胎便,可反覆發生尿道炎、陰莖頭炎和上尿路感染。
(二)中間位畸形約占15%。無瘺者直腸盲端位於尿道球部海綿體肌旁或陰道下段附近,恥骨直腸肌包繞直腸遠端。有瘺者其瘺管開口於尿道球部、陰道下段或前庭部。其肛門部位的外觀與高位畸形相似,也可以從尿道或陰道排便。探針可以通過瘺口進入直腸,在會陰部可觸到探針的頂端。在女孩以直腸前庭瘺多見,因瘺口位於陰道前庭舟狀窩部,也稱舟狀窩瘺,瘺孔較大,嬰兒早期通過瘺孔基本能維持正常排便,可引起陰道炎或上行性感染。
 |  |
診斷鑑別
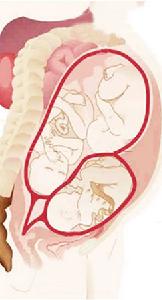 肛門直腸先天畸形
肛門直腸先天畸形(一)倒立側位X線平片稱為Wangenst-een-Rice法,要求在生後12小時以上攝片,等待氣體到達直腸,生活能力差者需要更長時間。在會陰肛門區皮膚上塗鋇劑作為標記,攝片前將嬰兒倒立2~3分鐘,使直腸盲端的胎便與腸管氣體互相轉換,採取髖關節呈90°屈曲位,使保持能充分顯示P點(恥骨中心)、C點(骶尾關節)、I點(坐骨最低點)的角度,以股骨大粗隆為中心,在呼氣、吸氣及啼哭時各攝片1張。
通過I點設一與PC線相平行的I線,與PC線間的距離為肛提肌群,直腸盲端位於PC線上方者為高位,於二線之間為中間位,超越I線為低位。或者設定M點,即坐骨結節的上2/3與下1/3交接點,在M線上方者為中間位,M線下方者為低位。
但必須注意各種影響因素,如腸道充氣不足、胎便過於粘稠,肛提肌的運動、X線投照角偏斜等均能影響位置的正確性。
(二)瘺管造影瘺管造影要求顯示造影劑注入時的結腸影象及造影劑排出時的直腸瘺管影象。結腸直腸與尿道雙重造影可顯示直腸瘺管與尿道的關係。陰道造影可顯示陰道與直腸的關係。
治療
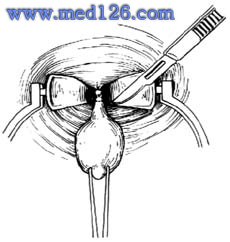 肛門直腸先天畸形
肛門直腸先天畸形後矢狀切口的手術方法,分離到直腸盲端,在直視下處理瘺管,以電刺激識別肌群的位置,保存直腸及肛周的肌肉神經血管組織,並恢復原狀,如若直腸太短或太寬,則從腹腔游離及作尾狀修剪,使直腸盲端準確通過肛提肌及括約肌群中央,從而得到滿意的排便功能。
低位畸形的治療,如肛門皮膚瘺無狹窄,排便功能無障礙者,不需治療。肛門或直腸下端輕度狹窄,一般採用擴張術多能恢復正常功能。對肛門皮膚瘺者,僅作簡單的"後切"手術。膜性肛門在新生兒期施行會陰肛門成形術。肛門前庭瘺如瘺孔較大,在一段時間內尚能維持正常排便,可於6個月以後施行手術。低位者因已通過恥骨直腸肌環,故手術較為容易,且術後排便功能良好。
泄殖腔畸形的治療,因一穴肛畸形複雜、新生兒期先作暫時性結腸造瘺,6月~1歲時行根治術。作成皮管或帶蒂小腸移植的陰道成形術,在新陰道後方行腹會陰肛門成形術,利用原泄殖腔構成尿道一部分,進行泌尿系器官成形術,爭取一期完成。
消化系統疾病
| 消化系統疾病一直是影響人類生存質量的主要疾病,特別是消化系統腫瘤更是嚴重威脅人類健康。醫學統計數據表明,屬於消化系統腫瘤的肝癌和胃癌長年來一直位於我國腫瘤發病率的第二位和第三位。 | |||
| 胃炎 急性胃炎 急性胃腸炎 胃潰瘍 慢性萎縮性胃炎 慢性胃炎 慢性淺表性胃炎 消化性潰瘍 胃腸道脹氣 腸上皮化生 胃癌 胃痛 急性胃擴張 胃柿石症 胃黏膜脫垂症 胃下垂 胃泌素瘤 傾倒綜合徵 胃扭轉 小腸腫瘤 阿狄森氏病 | 大腸癌 直腸癌 肛門直腸損傷 結腸直腸腺瘤 克隆氏病 腸中風 急性腸炎 慢性腸炎 急性腸梗阻 急性腸胃炎 偽膜性腸炎 放射性腸炎 直腸陰道瘺 腸結核 缺血性腸病 闌尾炎 急性闌尾炎 蛔蟲病 蟯蟲病 鉤蟲病 | 先天性肛門閉鎖 肛門瘺管 肛裂 肛門梳硬結 肛門濕疹 肛門直腸先天畸形 直腸脫垂 肛門瘙癢 大便失禁 肛管直腸癌 便秘 器質性便秘 功能性便秘 結腸性便秘 妊娠便秘 產褥期便秘 小兒便秘 急性細菌性痢疾 小兒便血 胃原性腹瀉 | 感染性腹瀉 食物中毒 腺病毒胃腸炎 慢性肝炎 小兒肝炎 藥物性肝炎 酒精性肝炎 肝昏迷 肝功能衰竭 肝細胞凋亡 肝腫大 黃疸 老年肝硬化 肝纖維化 門靜脈高壓症 肝腎綜合徵 肝吸蟲病 肝功能檢查 肝穿刺 肝移植術 |
