
疾病概述
 骨纖維異常增殖症
骨纖維異常增殖症骨纖維異常增殖症又名纖維性骨炎,是一種以骨纖維變性為特點的骨胳系統疾病,是否為一真性腫瘤尚無定論。該病好友於兒童及青年,女性較多見,主要有三種類型,①多骨型骨纖維異常增殖症;多發於四肢長骨,也伴發於扁平骨(顱骨、骨盆、肋骨等),常多處骨質受累,②單骨型骨纖維異常增殖症;多發生於顱面骨,以上頜骨多見,在臨床上該型與耳鼻咽喉科關係密切,常被誤診為上頃塞惡性腫瘤。③艾布賴特綜合徵(Albright’ssyndrome):由多骨型骨纖維異常增殖(稱播散性纖維性骨炎)、皮膚色素沉著及內分泌障礙(以女子性早熟為突出表現)等症狀構成。骨纖維異常增殖症為緩慢進行性局部腫塊,因腫塊壓迫鄰近器官組織,產生各種機能障礙與畸形,
本病尤其是單骨型,主要以手術切除為主,因放療有誘發惡變可能。鑒於本病臨床進展緩慢,對病變較小或無症狀者,可暫不手術,但應密切隨訪觀察。病變發展較快者,伴有明顯畸形和功能障礙者,應視為手術指征。根治性切除雖為最佳治療方法,但有導致功能障礙與美容缺陷之弊。保守的部分切除易於復發,其中單骨型為21%,多骨型可高達36%。手術方法和進路選擇,應根據原發部位、侵犯範圍和功能損害程度靈活掌握,原則上是儘可能徹底清除病變組織,又能最大限度地保留器官生理功能和美容效果。
疾病原因
1病因學
 骨纖維異常增殖症
骨纖維異常增殖症2病理改變
血管供應變異較大,病變組織大體呈白色、灰白色或蒼黃色,比正常骨組織稍軟,切割時有含砂感或彈性感,巨大骨損害多從骨髓向外侵蝕和擴展,管狀骨和扁平骨的骨皮質僅留兩層薄殼,去除外殼如去包膜。鏡下見:網狀骨骨小梁的大小、形狀和分布不一,無規律地包埋於質地疏鬆或緻密的富含細胞和血管的結締組織中。此組織類似結締組織化生的結果。骨小梁形態變異較大,多呈球形,在橫切面呈曲線形、C形或弓形,邊緣不規則,骨細胞腔隙寬闊。骨小梁緊密排列,形成骨網。骨小梁由粗纖維的原骨構成,在偏振光鏡下呈網狀而非板狀。偶見網狀骨板狀變形,有時見弓狀骨小梁環繞一中心血管。多數骨小梁缺乏成骨細胞構成的輪廓。這可與骨化纖維瘤鑑別
疾病表現
 骨纖維異常增殖症
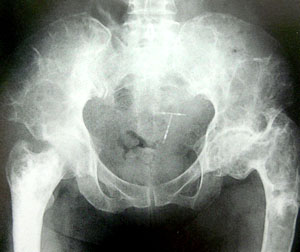
骨纖維異常增殖症臨床分類-即將本病分為三型:①單骨型:單個或多個損害累及一塊骨,其中上頜骨發病最多,為64%,下頜骨為36%,顱面骨為10%。②多骨型但不伴內分泌紊亂:多個損害累及一塊以上骨骼。在中等度骨骼受累的多骨型中,顱面骨受累的發生率為5%,在骨骼廣泛受累的多骨型中,顱面骨受累的發生率為100%。單骨型與多骨型在顱面骨中以額骨和蝶骨受累者最多,且常同時受累,其次為篩骨和顳骨。可單側或雙側同時發病。③多骨型伴有內分泌紊亂:此型與單骨型比例為30∶1。損害散布於多個骨骼,常為單側分布,伴有較大皮膚色素斑。多見於女性,表現第二性徵早熟。
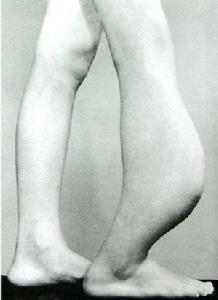
臨床表現本病約60%發生於20歲以前,偶見於嬰兒和70歲以上老年人。男女發病為1∶2。80%以上表現為病骨區畸形腫脹,發生於面部者表現兩側不對稱,眼球移位、突出,鼻腔狹窄,牙齒鬆動,齒槽嵴畸形,流淚,齶部隆起。隨著病變發展可出現頭痛和偶爾發生鼻衄。由於原發部位和累及的範圍不同,可表現出相應的臨床症狀。如發生於顳骨,常表現顳骨體積膨大變形,外耳道狹窄,傳導性耳聾。有外耳道狹窄者,約16%並發膽脂瘤。有膽脂瘤者,常導致顳頜關節炎、面癱、迷路炎或顱內併發症,病變累及耳蝸及內聽道可產生感音性耳聾。岩骨受侵,易出現顱中窩或顱後窩受累症狀。此病可廣泛侵入鼻竇、眼眶及顱前窩底,臨床呈惡性生長傾向,表現為鼻塞、嗅覺減退、面部不對稱、眼球突出、移位、復視、視力障礙和張口困難等。蝶骨和蝶竇區骨纖維異常增殖,多有較嚴重的額頂或枕區疼痛。由於蝶竇壁菲薄,病損易向周圍結構擴展,累及Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ等顱神而產生顱神經受損症狀與體徵。病變較大者可致腦萎縮或產生高顱壓症。
2、影像學表現
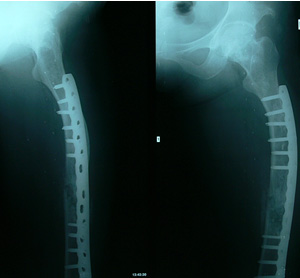
一、病灶多發生於四肢長骨,尤以股骨和脛骨為多見,其次為肋骨、顱面骨、骨盆和手、足小骨,而脊柱較為少見。
二、X線上,病灶的密度常有差異,取決於病理成分,病灶如主要為纖維組織常表現為囊狀透光區;如主要為砂礫樣鈣化新生骨者常呈磨砂玻璃狀;如新生骨鈣化較多時則表現為一片明顯的增白區。每一病灶,可以上述表現按不同比例組合出現。
 骨纖維異常增殖症
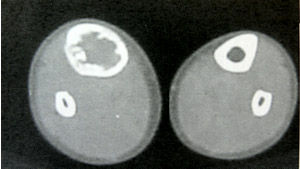
骨纖維異常增殖症1、囊性
(1)囊性:以四肢骨多見。
(2)表現為囊狀透光區,皮質變薄,骨幹可有膨脹,囊內有磨玻璃樣改變及鈣化。
(3)單囊可發展成多囊,囊內有粗大的骨小梁,邊緣可有硬化 。
2、硬化型
(1)以顱面骨和顱底骨多見。
(2)表現為瘤骨密度非一致性增高,在硬化區內有散在的顆粒狀透亮區。
(3)顱骨穹隆的病。變常侵犯外板和板障,使骨骼膨大、增厚和囊性改變,呈磨玻璃樣或緻密硬化改變。
(4)面骨主要侵犯上頜骨,硬化區波及顳骨及眶下緣,並占據上頜竇腔。
3、位於長、短管狀骨和肋骨的病灶多發生於骨幹或骨骺端,病骨膨脹而變粗大,常呈單房透明或磨砂玻璃狀,範圍較大,其中可有緻密骨嵴沿骨長軸方向走行向內凸出,使病灶呈不完全的分房狀如“絲瓜囊”。有時在病灶內可見或大或小的片狀鈣化影。
4、位於顱底骨和面骨的病灶以硬化型多見,表現為骨密度均勻增高,骨質增厚,與正常骨分界可清楚不清。
疾病檢查
 骨纖維異常增殖症
骨纖維異常增殖症①變形性骨炎型:常為多骨型病變表現,其特點是顱骨增厚,顱骨外板和頂骨呈單側泡狀膨大,骨內板向板障和顱腔膨入,增厚的顱骨中常見局限和瀰漫的射線透明區和濃密區並存,這種骨吸收與硬化並存極似Paget變形性骨炎的表現。顱骨擴大和硬化,可從額骨擴大到枕骨。面部受累可導致眶和鼻腔狹窄及鼻竇腔消失,此型約占56%。②硬化型:此型多見上頜肥厚,可致牙齒排列不整,鼻腔、鼻竇受壓變小。上頜骨受累多於下頜骨,且多為單骨型。損害呈硬化或毛玻璃樣外觀。相反,下頜骨損害多見於多骨型,表現為孤立的骨壁光滑且可透過射線。此型約占23%。③囊型:顱骨呈孤立或多發的環形或玫瑰花形缺損,缺損從菲薄的硬化緣開始,其直徑可達數厘米。孤立的損害有似嗜酸性肉芽腫,多發的缺損可誤認為HandSchüllerChristian病,偶有數種X線類型出現於同一個體上。此型約占21%。套用CT或MRI檢查,能明確病變的位置和範圍,且能顯示與軟組織的聯繫。定期檢查可動態觀察病變的發展程度,對選擇術式進路、減少併發症和估計預後甚為重要。
鑑別診斷
在診斷過程中應注意與下列疾病鑑別:
 骨纖維異常增殖症
骨纖維異常增殖症2.嗜酸性肉芽腫為一良性孤立的非腫瘤性溶骨損害,起源於網狀內皮系統。常見於額骨、頂骨和下頜骨。多發於30歲以前,男性居多。在組織學上,由濃密的泡沫組織細胞組成,伴有不同數量的嗜伊紅細胞和多核巨細胞。組織細胞核含有小囊,嗜伊紅細胞含有細小的空泡,巨細胞為郎罕型和異物型。這些細胞呈灶性集聚。
3.Gardner綜合徵此綜合徵為侵犯上下頜骨、顱骨和偶見於長骨的多發性骨瘤,伴有腸息肉、皮樣囊腫、纖維瘤和長骨局灶性波紋狀骨皮質增厚。
4.巨型牙骨質瘤通常累及下頜骨全部,可致骨皮質膨大,X線檢查表現為濃密的塊狀堆積體。常起於遺傳,在組織學上未發現感染源。
5.外生性骨瘤副鼻竇惡性腫瘤及囊腫等,均應注意鑑別,以防誤診。
6.多骨型骨纖維異常增殖症,還應與甲狀腺功能亢進、paget病、神經纖維瘤病及頜骨肥,症等相鑑別。
疾病治療
 骨纖維異常增殖症
骨纖維異常增殖症手術中,對兒童及病變廣泛者,宜採用經口插管全麻。病變局限者亦可在局麻下切除。
手術切口有多種,可以根據病變情況選擇套用。①Caldwell-Luc法:適用於病變廣泛累及上頜骨、鼻腔、眶下壁、篩竇和蝶竇者;②Weber-Fergusson法:適用於上頜骨、眶下壁、顴骨、硬齶及蝶篩竇廣泛受侵的病人。③顱—面聯合進路:包括雙額瓣或單額瓣+Weber—Fergusson切口,適用於原發於顱前窩底或鼻竇、眶壁並互為侵犯的廣泛病變。④Fish法:適用於病變原發於顳骨,外耳道、中耳、內耳、岩骨及顱中窩底受累者。
手術以平鑿、圓鑿或大刮匙分次切除為好。創面滲血較多,術中宜使用骨蠟止血和給予必要的輸血,尤其是兒童更應注意。
手術擬分兩期進行。把兩期手術要點分別概括為“換頭修面”和“換臉整容”。
第一期手術將把病變的頭蓋骨全部切除,換成鈦合金的頭蓋骨。因為患者現在的頭蓋骨骨纖維帶有病變,單純把增生的頭骨削薄繼續使用,術後很可能會再次增生。此外還要進行頜面部異常骨結構的切除與整復,施行顱眶重建,將突出的眼球回納,整復畸形外鼻和口唇以及頭面部皮膚整形。通過第一期手術,患者五官將基本恢復正常。
第二期手術重點解決面部皮膚顏色不一致的問題,切除面部病變的皮膚,取自體帶蒂皮瓣移植,對面部分區整容,使患者面部皮膚顏色基本達到一致。本病手術切除預後良好,故術中對鄰接顱底及顱內的重要神經、血管部位病變,不要過份切除,以免發生意外。
普通治療多採取病段切除後行帶血管的骨瓣移植、搔刮後植骨等治療。但是植骨後由於各種因素影響植骨存活,新植骨與囊壁的融合連線困難,難以修復。多骨型伴病理性骨折治療就更為困難。中藥治療骨纖維異常增殖症主要是調理內分泌,改善重建微循環,激活成細胞生長。它能使囊腔和瀰漫的骨溶解區逐漸修復,骨小梁由稀疏變為緻密,骨皮質逐漸增厚。最新套用Ilizarov技術,病灶段切除,自正常骨組織處延長再生能較快的徹底治療骨纖維異常增殖症。
疾病預防
本病目前病因不明,尚無有效的預防措施,故早診斷早治療是本病的防治關鍵。
