
簡介
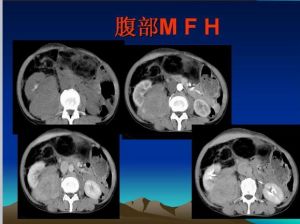 惡性纖維組織細胞瘤影像學
惡性纖維組織細胞瘤影像學惡性纖維組織細胞瘤(Malignantfibroushistiocytoma,簡稱MFH)是中老年最常見的惡性腫瘤之一,這種間質起源的肉瘤雖可發生於身體的任何器官,但常侵犯四肢及腹膜後。1972年Feldman和Norman提出將此瘤作為一種獨立的骨腫瘤類型,以前常將骨內MFH誤認為骨肉瘤、纖維肉瘤、骨巨細胞瘤或骨轉移性癌等。
惡性纖維組織細胞瘤由可分化為組織細胞和成纖維細胞的細胞形成。近年來,已發現此腫瘤的變種有:多形分層型,粘液型,巨細胞型、炎症型、血管瘤樣型等。除了代表其各自不同的解剖—臨床病種和不同的治療方法和預後外,這些變種均應被認為是屬於惡性纖維組織細胞瘤的臨床表現中的一部分。同時,這些變種特有的症狀往往可以做為確定診斷的重要依據。
本病不少見。好發於男性,多見於成人和老年人。常位於長骨,依次為股骨、脛骨和肱骨,象骨肉瘤一樣,較常發生於股骨遠端和脛骨近端,與骨肉瘤不同的是此病更容易從乾骺嘀蜮骨幹—乾骺端向骨幹侵犯,由於患者一般是成人,因此可侵及骨骺。有時可僅發生於骨幹,或僅在短骨和扁平骨見到。一般在患者就診時,症狀(疼痛和腫脹)出現的時間很短,但有時可在1~2年以上。
本病又稱為纖維黃色肉瘤,是一種多形性肉瘤,可能為中老年最常見的軟組織肉瘤。有的病例以前診斷為多形性脂肪肉瘤、纖維肉瘤或橫紋肌肉瘤,現在均屬於本病。腫瘤大小,特別是腫瘤深度,與存活率關係很大。
此瘤為具有多形性的高度細胞性腫瘤,其中有些細胞,由於儲有脂質,空泡化很明顯,核絲分裂象常見,有些呈非典型性。細胞Vimentin染色陽性。手術切除後復發率為25%,35%發生轉移,存活率為50%。
關於最新命名
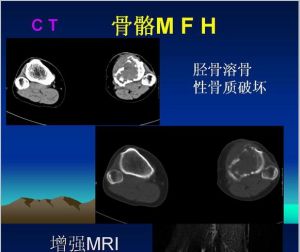 惡性纖維組織細胞瘤影像學
惡性纖維組織細胞瘤影像學儘管MFH給人以纖維母和組織細胞起源的印象,其確實起源爭論了幾十年。多種技術(包括細胞培養、免疫組化、電鏡)和研究試圖闡明其組織起源,然而不同的研究結果互相矛盾,其差異性令人吃驚。組織細胞、纖維母、介於二者之間的中間細胞都曾被提出為腫瘤細胞起源。後來提出一種具有一定程度的兼有纖維母和組織細胞分化的原始間葉性幹細胞。這些研究的大量證據表明MFH為纖維母或原始間葉起源的肉瘤,表現為兼有纖維母和組織細胞分化的特徵,而不是真性組織細胞起源。然而,在最終明確腫瘤細胞起源之前,2002年版WHO軟組織腫瘤分類仍將它保留在“所謂纖維組織細胞性”大類中。
現在認為MFH是一種共同的“形態結構”,見於多種多形性腫瘤,而不論何種分化。因此,MFH將實際上無關但有相似形態特徵的不同腫瘤聯繫起來。MFH也被認為代表腫瘤生長的一種“最後共同通路”。因腫瘤(肉瘤或非肉瘤)在生長過程中的進展,它們可能失去分化結構而最後形成未分化結構。一些研究重新評價了原診斷為MFH的病例,結果支持這些假說,因這些病例可能查出分化(脂肪源性、神經源性、肌源性或非肉瘤性)。這可以解釋:(1)許多診斷為MFH的腫瘤在免疫組化、超微結構和細胞遺傳學方面的異質性;(2)臨床行為以及結局的寬廣譜系(包括治療敏感性、轉移時間和方式);(3)少見部位頻繁出現MFH樣病變。與此相似,MFH亞型的形態特徵也是許多其它腫瘤共有的,使診斷分類(實體)產生疑問,難以歸類。鑒於此觀點,MFH及其亞型的診斷變為局限於一小部分多形性肉瘤,它們被排除了所有其它可能的分化譜系。
現在認為MFH這一術語是錯誤的名稱(誤稱),因為它提出的組織細胞起源這個觀點已經沒有根據,而且它錯誤地包括了具有共同形態的不相關腫瘤,如前所述。因此現在強烈推薦摒棄這個名稱。WHO提倡用“多形性肉瘤(pleomorphicsarcoma)”取代它,給予腫瘤更準確的形態學描述而且不提示腫瘤細胞的起源。也有提議用“多形性纖維肉瘤(pleomorphicfibrosarcoma)”一詞,但反對意見認為,它可能與經典的纖維肉瘤(classicalfibrosarcomas)混淆,後者是不同實體,由相對一致的梭形細胞群組成,並且幾乎總是沒有多核或多形性巨細胞。不幸的是,MFH這個名稱已經深入外科病理並且為外科醫生和臨床所熟知。因此,推薦在使用新命名時,同時列出MFH的舊名稱。2002年版WHO軟組織腫瘤分類用這種方式保留了MFH這個名稱。2002年版美國腫瘤聯會(AmericanJointCommitteeonCancer)出版的有關軟組織腫瘤的分期系統也使用了相同的方式。
其分類
 惡性纖維組織細胞瘤影像學
惡性纖維組織細胞瘤影像學歷史上提出過五個MFH亞型:(1)車輻狀-多形性MFH(storiform-pleomorphicMFH,WHO稱為pleomorphicMFH多形性MFH);(2)粘液樣MFH;(3)炎症型MFH;(4)巨細胞型MFH;(5)血管瘤樣MF(angiomatoidMFH)。現在已澄清,這些亞型為異質性實體,不應歸入作為單一分類。
車輻狀-多形性MFH在MFH及其亞型作為診斷實體被推翻之前,車輻狀-多形性MFH占所有MFH病例的絕大多數(60%-70%)。典型的車輻狀-多形性MFH由梭形細胞夾以多角形或圓形細胞混合組成,排列成車輻狀結構。也可出現數量不等的奇異形、多核巨細胞。細胞密集,核多形性明顯,伴較多非典型性核分裂像。車輻狀-多形性MFH通常見於老年(60-70歲)患者的下肢,後腹膜也是常見部位。前述“共同結構/最後通路假說”可很清楚地解釋車輻狀-多形性MFH的形態特徵:腫瘤形態為許多未分化腫瘤所共有,包括肉瘤和非肉瘤性腫瘤,後者如未分化癌、梭形細胞惡黑、梭形細胞淋巴瘤。因此,車輻狀-多形性MFH的診斷是一種排除性診斷,在無法識別分化譜系時才能診斷。在這種情形下,WHO2002提倡用“未分化高級別多形性肉瘤”取代它,但仍將它放在纖維組織細胞腫瘤一類之下。隨著概念轉變,一度認為此亞型為成人最常見的軟組織腫瘤,現在發現不超過成人STT的5%。
粘液樣MFH粘液樣MFH是第二常見亞型(10-20%)。鏡下,腫瘤有明顯的粘液樣基質。現在認為粘液樣MFH為一個特殊實體,不僅由於其獨特的粘液樣外觀,而且它比其它亞型有更好預後。因此WHO2002使用粘液纖維肉瘤這個術語。因粘液纖維肉瘤表現為肌源性分化(SMA+或MSA+),它從纖維組織類轉移到肌纖維母細胞類。
巨細胞型MFH巨細胞型MFH為罕見亞型(10-15%)。鏡下特徵是多核巨細胞。巨細胞很像骨母細胞,但它們傾向於有更高級別的核,很少發現與類骨質(osteoid)有關。現在認為巨細胞型MFH不代表一類特殊實體,因為其中的大部分通常可能識別出分化譜系。許多原診斷為巨細胞型MFH的病例現在被重新分類為巨細胞豐富的骨肉瘤、平滑肌肉瘤伴骨母細胞樣巨細胞反應,或巨細胞豐富的間變性癌。如果未發現分化證據,可診斷為巨細胞型MFH,並使用新名詞—伴有巨細胞分化的未分化多形性肉瘤。在2002版WHO軟組織腫瘤分類中,它被歸入纖維組織細胞大類下。
炎症型MFH炎症型MFH為最少見亞型(5%)。特徵為緻密炎細胞浸潤(中性粒細胞、淋巴細胞、泡沫細胞為主,圖5)。與其它亞型相似,此亞型也被質疑。許多原診斷為炎症型MFH的病例被重新診斷為去分化脂肪肉瘤,其去分化成分有明顯間質炎細胞浸潤。其它與炎細胞浸潤非常相似的病變包括間變性癌伴明顯炎症和間變性大細胞淋巴瘤。因此,炎症型MFH的診斷僅能在無分化譜系時成立。在2002版WHO軟組織腫瘤分類中,它被改名為伴有明顯炎症的未分化多形性肉瘤,放在纖維組織細胞腫瘤大類中。
MFH亞型的舊名稱
MFH亞型的新名稱(編碼)
腫瘤分類
(編織狀-)多形性MFH
未分化高級別多形性肉瘤
所謂纖維組織細胞性
粘液樣MFH
粘液纖維肉瘤
肌纖維母細胞性
巨細胞型MFH
伴有巨細胞分化的未分化多形性肉瘤
所謂纖維組織細胞性
炎症型MFH
伴有明顯炎症的未分化多形性肉瘤
所謂纖維組織細胞性
血管瘤樣MFH
血管瘤樣纖維組織細胞瘤
分化不確定的腫瘤
血管瘤樣MFH血管瘤樣MFH現在認為不是MFH亞型,因形態和臨床都與MFH及其亞型不同。血管瘤樣MFH主要見於兒童和年輕成人。鏡下,腫瘤良性外觀,卵圓形、圓形或梭形細胞,嗜酸性,輕微多形性,排列成片或鏇渦。有明顯的裂隙樣血管腔,伴細胞浸潤、局灶含鐵血黃素沉著和出血。而且,腫瘤傾向於惰性,極少轉移。其新名詞血管瘤樣纖維組織細胞瘤(angiomatoidfibroushistiocytoma,AFH)可反應良性性質。儘管一半病例表現為Desmin+並反應其肌源性分化,其確切的分化譜系仍然不明,因為常常同時出現EMA+提示上皮性分化。因此,AFH從纖維組織細胞性分類中移出,放入分化不確定的腫瘤中。
發病誘因
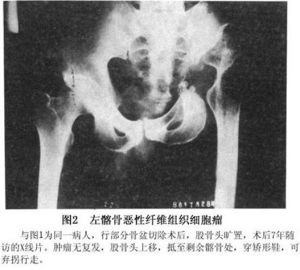 惡性纖維組織細胞瘤影像學
惡性纖維組織細胞瘤影像學(一)發病原因
大多數惡性纖維組織細胞瘤的病因不明,最近的免疫組織化學技術已有一些證據表明惡性纖維組織細胞瘤與纖維母細胞在表現上關係密切。大約半數的骨內惡性纖維組織細胞瘤與其他病變的發生有關係:如骨梗死、Paget病、纖維異樣增殖症等,放射線可以誘導產生惡性纖維組織細胞瘤。
惡性纖維組織細胞瘤,可以是原發的,也可以是繼發於骨梗死、Paget病或是放射治療之後。這表明骨壞死或良性的慢性修復過程中,豐富的單核和多核組織細胞和纖維母細胞的增殖是造成繼發惡性腫瘤的原因。
(二)發病機制
肉眼觀察腫瘤在骨內破壞廣泛,晚期侵蝕骨皮質累及鄰近軟組織,瘤體本身呈灰白色,魚肉狀,可因出血壞死的不同程度呈各種顏色。鏡下所見主要成分有兩種即腫瘤性成纖維細胞與間變的組織細胞,前者為梭形細胞,肥碩梭形,細胞間有數量不等的膠原纖維,細胞有不同程度異形性及核分裂相,細胞排列緻密。
常可見具特徵性的花瓣狀(storiformpattern)。組織細胞較肥大,呈圓形或卵圓形,胞質豐富嗜酸性,有吞噬功能,吞食脂類物質,胞質內可呈顆粒狀、泡沫狀或空泡狀,細胞核圓形、卵圓形、腎形或多邊分葉狀,核仁清楚,組織細胞的異形性十分明顯,核分裂多見,其他成分可見瘤巨細胞、多核瘤巨細胞、Touton巨細胞、黃色瘤細胞及組織壞死、鈣化、黏液變性、膠原透明變性、淋巴細胞浸潤和合併血管瘤成分等。
MFH的電鏡所見纖維母細胞呈卵圓形或梭形,胞質突起,含有大量溶酶體,並有高爾基器,粗面內質網及線粒體,分化差者就較少,細胞核圓形、卵圓形或不規則形,常有雙核,核膜下有大量異染色質。組織細胞為卵圓形或不規則形,細胞表面伸出偽足,有絨毛狀皺褶,還有高爾基器、粗面內質網及線粒體,並有多量脂滴及糖原顆粒。
臨床表現
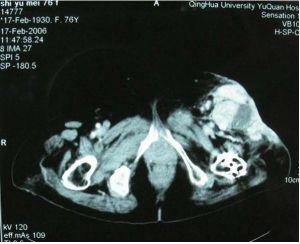 惡性纖維組織細胞瘤影像學
惡性纖維組織細胞瘤影像學最常見到的軟組織惡性腫瘤之一。由於近年來將以往單獨分類為我形性橫紋肌肉瘤、多形性脂肪肉瘤和罕見的稀疏分化性纖維肉瘤等均歸屬於惡性纖維組織細胞瘤,所以,後者的發病數量增多,範圍擴大。
根據臨床工作需要,又將上述病變分為數種變種。通常,在對無特殊細胞分化改變的多形性肉瘤重複進行檢查時,即顯示惡性纖維組織細胞瘤的形態特徵。惡性纖維組織細胞瘤明顯好發於男性。
發病年齡較晚,一般從50~70歲。僅血管瘤樣型者可在20歲以前發病。惡性纖維組織細胞瘤好發於肢體(特別是下肢、尤以大腿為多見)及腹膜後者主要為炎症型變種。約90%以上病變的部位較深,多在筋膜下發病,約10%的病變發生在淺表部位。相反,血管瘤樣型(變種)者好發於肢體的皮膚和皮下組織。
為在深層逐漸生長的球形腫塊。有時生長較緩慢,有時則非常快。一般無疼痛症狀。通常從發生腫脹至確定診斷的時間,從數月到數年不等。位於腹膜後者診斷較難和較遲。症狀包括厭食、體重下降及腹腔器官受壓等。對位於筋膜上的病變較易診斷,當腫瘤形體為中等大小時即可發現。有時腫瘤呈囊性變和/或出血,以致有可能誤診為血腫。
影像學表現
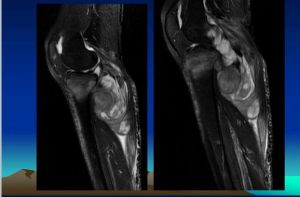 惡性纖維組織細胞瘤影像學
惡性纖維組織細胞瘤影像學少數情況下,當腫瘤貼近骨骼時,可能顯示淺表性骨質溶解或骨膜反應。骨掃描檢查對鄰近的骨反應很敏感。很少見到病灶的鈣化和骨化,但有時也可在腫瘤周圍或腫瘤內見到。
血管造影可顯示其與其他惡性腫瘤所共有的變化。但有時甚至在非常大的腫瘤內,可因組織壞死和出血,而呈現為無血管的影像。主要血管可能移位和受壓,但很少被浸潤。CT和MRI顯示為實心的,非均質性腫塊。有時可見大的含有液體的囊腔。
病灶呈灶樣缺損的透亮區,多有完整邊緣,好發於乾骺端,經常可見有皮質破壞,並穿透到軟組織中去。無明顯骨膜反應。X線徵象類似於其他原發或繼發的惡性腫瘤,無特徵性,全身骨掃描和CT檢查有助於腫瘤鑑別的來源為髓內或骨外。
輔助檢查
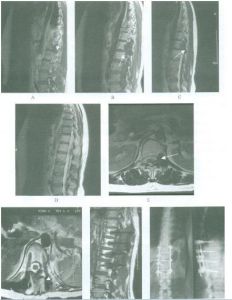 溶骨性侵襲性破壞,軟組織腫塊較明顯,少有骨膜反應和鈣化
溶骨性侵襲性破壞,軟組織腫塊較明顯,少有骨膜反應和鈣化1、X線檢查 軟組織惡性纖維組織細胞瘤病變表現為密度均勻一致的腫塊,內無鈣化及骨化。骨內惡性纖維組織細胞瘤呈形式多樣的骨破壞:或為大片狀的溶骨性骨破壞,邊緣模糊不清,邊緣有時伴有篩孔樣骨破壞;或為類圓形的單囊、多囊狀骨破壞,周邊有硬化環包繞。本病一般無骨膜反應,皮質破壞者可伴有軟組織腫塊。
2、CT檢查軟組織惡性纖維組織細胞瘤CT圖像表現為密度均一的腫塊。骨內惡性纖維組織細胞瘤CT影像的差異較大,可以觀察到腫塊自骨破壞中心向四周組織中蔓延,多呈分葉狀,無完整包膜,軟組織腫塊有時巨大,呈低密度浸潤狀生長,邊緣不清,腫物中心常有壞死囊變區。
3、MRI檢查多數惡性纖維組織細胞瘤的邊界清楚,少數呈侵潤性生長,邊界不清楚,腫瘤呈結節狀,可有分葉。在T1加權像為低或中等信號,T2加權像呈高信號,注射Gd-DTPA增強後,腫瘤內不均勻強化。
4、病理檢查
肉眼所見軟組織惡性纖維組織細胞瘤的大體特徵同軟組織肉瘤,骨內惡性纖維組織細胞瘤與軟組織惡性纖維組織細胞瘤外觀相同。
鏡下所見惡性纖維組織細胞瘤的典型特徵是:1、惡性梭形細胞(即纖維細胞)呈“輪輻狀”排列,構成腫瘤的基質;2、有的區域內含有體積巨大形態奇特的組織細胞,細胞內含有大量頗具特徵的嗜酸性胞漿;3、大量的有絲分裂象;4、散在的慢性炎症細胞。上述特點出現的多少可以因病變而異,常見的主要特徵是纖維成份以及形態奇特的惡性組織細胞。
診斷要點
1、軟組織的MFH表現為侵襲性增大、位置深在但症狀非常輕微的軟組織腫塊。
骨的MFH多發生於主要長骨且呈侵襲性生長,因而經常出現病理骨折。
2、X線表現軟組織MFH沒有獨特的X線徵象籍以與其他軟組織肉瘤相鑑別。病變內鮮有鈣化或/和骨化區,也沒有X線密度增高或減低區,因此無法提示病變的組織發生。骨內MFH呈穿透樣密度減低區,邊緣不清,皮質破壞並伴有軟組織腫塊。
診斷進展
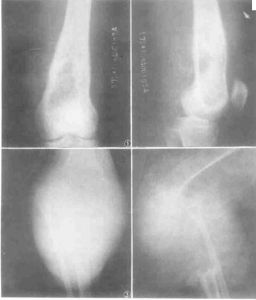 惡性纖維組織細胞瘤診斷
惡性纖維組織細胞瘤診斷儘管強調盡力尋找分化譜系,目前尚無診斷進展的指南。然而,仔細和/或廣泛取材,並使用輔助技術特別是免疫組化,應該是兩個重要原則。
仔細和/或廣泛取材任何類似MFH的腫瘤的診斷進展,都應該從仔細取材開始,以尋找有助於識別分化譜系的區域。如果最初取材的切片對識別分化譜系沒有幫助,再次取材可能有助於發現較分化區域,或至少能發現一些初次取材未見的診斷特徵。一個研究中,3個後腹膜的去分化脂肪肉瘤最初都被診斷為MFH,這是因為取材不夠。
免疫組化MFH樣腫瘤的診斷,現在強調免疫組化的重要性,單獨依據形態學診斷的MFH已經不被承認。因MFH的診斷是排除性的,可能需要廣泛使用一組免疫組化,以排除不同的分化譜系。現在沒有一個明確指南規定免疫標記物應該有多廣泛,但問題應該尋求免疫標記物的正確使用。免疫標記物的組合,應該從廣譜分化開始,如VIM用於間葉分化,CK用於上皮分化。然後根據臨床和組織學所懷疑的方向,增加更特異標記物。典型的未分化多形性肉瘤呈VIM+而其它分化譜系的標記陰性。與傳統認識相反,組織細胞標記物(CD68,α1-antitrypsin抗胰蛋白酶,α1-antichymotrypsin抗胰蛋白酶,XIII因子)不再有助於MFH的診斷,因為這些標記物無特異性,不能支持MFH的明確診斷。
電鏡組織形態學和免疫組化不足以作出特異性診斷時,電鏡可能有助於尋找MFH樣腫瘤的分化譜系。免疫組化不能識別腫瘤時,電鏡可能提供答案,特別是腫瘤表達小灶明顯異常標記物時。例如,未分化多形性肉瘤灶性表達CK或散在SMA的現象並不少見,不要認為是癌或肌源性分化的證據。此時,電鏡可以通過發現特徵性超微結構生物以證實分化譜系。分子技術大量研究報導了MFH中的不同遺傳學異常,但明確可靠的遺傳學數據仍沒有,這限制了分子技術在診斷中的用途,至少現狀如此。
在進一步使用輔助技術之前,通常推薦仔細取材和ihc研究,以努力確診MFH。然而,這個診斷進展有局限性。它基於一個假設,即:有足夠量的組織供廣泛取材。這可能不現實,特別是穿針穿刺活檢,它限制了可用的組織量。而且,如果廣泛取材並使用大量IHC,診斷過程可能太費力並且太昂貴。因此,外科病理學家必須在有限的時間和費用之內,追求更準確和更有效率的診斷程式。必須制定準確、可重複的診斷指南,以幫助有邏輯性、有序地尋找MFH樣腫瘤的分化譜系。
MFH樣腫瘤尋找分化的意義因MFH形態為許多不同譜系的腫瘤所共有,識別其分化就有治療意義,因為這些腫瘤有不同的治療方式。例如,低分化腺癌、梭形細胞淋巴瘤和惡黑都可有MFH樣形態,但治療不同。如果僅根據形態學誤診為MFH,會誤導治療。
識別多形性肉瘤的亞型可能有預後意義。例如,有肌源性分化的多形性肉瘤(如多形性橫紋肌肉瘤)比無肌源性分化者(如多形性脂肪肉瘤)通常更侵襲性。
細胞學細針穿刺標本對鑑別診斷價值有限。細針穿刺標本的MFH的細胞學特徵無特異性,可見成簇不典型多角形和梭形細胞,伴奇異形、多核巨細胞。腫瘤細胞不顯示任何特異性組織分化的細胞形態學證據。因通常需要廣泛IHC,沒有理由僅根據細針標本期望準確分類,特別是如果細胞學標本量不足以做細胞塊供IHC。因此,最好避免僅根據細胞學表現診斷MFH。然而,努力尋找有助於顯示特異性分化的細胞或特徵(脂母細胞,橫紋肌母細胞,角化證據,粘液等),可以幫助排除MFH的診斷。
最後,我們必須指出,MFH診斷標準的變化已經影響文獻。這種改變可能影響到許多曾經報導的MFH的可靠性。病理學家應該慎重接受以往僅根據形態學診斷的MFH的正確性,特別是在IHC年代之前。而且,已出版的有關MFH治療方式和預後的臨床試驗也必須用新的限於不可分類的腫瘤—多形性肉瘤—的治療策略更新。
鑑別診斷
1、骨肉瘤好發年齡為10~20歲,臨床表現為疼痛、不斷增大的腫塊,血清鹼性磷酸酶可升高,X線表現為破壞性病灶,內有不規則的成骨,常見放射狀骨針和Codman三角。本病與骨肉瘤鑑別要點是:骨肉瘤常有明顯的骨膜反應及Codman三角,以及多種形態、不同密度的瘤骨,發病年齡以青少年多見。
2、纖維肉瘤其與惡性纖維組織細胞瘤在臨床和影像學上無明顯區別,其鑑別主要依據病理學檢查,前者的主要細胞學特徵是由大小、形態均一的梭形細胞構成,細胞核深染,幾乎沒有胞漿,細胞膜不明顯或缺如。細胞被膠原纖維間隔,交織排列,呈“鯡魚骨”狀。後者主要特徵是纖維細胞呈“輪輻狀”排列,構成腫瘤的基質,有的區域內含有體積巨大形態奇特的組織細胞。
疾病治療
 惡性纖維組織細胞瘤
惡性纖維組織細胞瘤1、非手術治療
(1)放療:對於軟組織惡性纖維組織細胞瘤,術前放療是一種非常有用的輔助療法,放療可使病變產生大量壞死,並且在大多數的病例中可刺激產生一層緻密的纖維包殼,從而使廣泛或者邊緣性切除的保肢手術更加安全。對於骨內惡性纖維組織細胞瘤術前放療作用有限。
(2)化療:骨內惡性纖維組織細胞瘤對化療的反應比不上骨肉瘤或淋巴瘤。多數學者認為惡性纖維組織細胞瘤術前術後輔以化療可以提高生存率。單純外科治療不能控制病灶,而接受外科治療與輔助化療的病人五年生存率明顯提高,常用的藥物有HD-MTX-CF、ADM和VCR(長春新鹼)。
2、手術治療
(1)廣泛切除:適應於I級惡性纖維組織細胞瘤;術前輔助治療反應滿意的II期腫瘤,也可行廣泛切除。骨內惡性纖維組織細胞瘤切除後根據部位可選擇適當的重建方法:如關節融合術、人工假體置換術、同種異體骨關節移植術、異體骨和人工假體複合移植術、帶血管自體骨移植術、瘤骨滅活再植術等。
(2)根治性切除:術前輔助治療反應不滿意的II期腫瘤可行根治性切除術。根治切除範圍應距瘤體3~5cm,若情況許可,應距瘤體儘量遠,如上範圍的擴大切除可使復發率從50%降至26%。
(3)截肢術:主要用於多次復發者,可明顯降低復發率,但遠處轉移率無明顯降低。
預後
變種的不同形態學外觀,對診斷特別重要。事實上,在變種和預後之間並無明顯的關係。後者似與臨床因素,如好發及所在部位、形體大小和腫塊的局限性等關係更為密切。例如,局限在皮下組織的腫瘤約10%可發生轉移,而與部位較深者相比,後者約40%發生轉移,在肢體中,遠側發病較近側者預後好。預後最差者為腹膜後腫瘤。
預防
一、變種的不同形態學外觀,對診斷特別重要。事實上,在變種和預後之間並無明顯的關係。二、後者似與臨床因素,如好發及所在部位、形體大小和腫塊的局限性等關係更為密切。
三、例如,局限在皮下組織的腫瘤約10%可發生轉移,而與部位較深者相比,後者約40%發生轉移,在肢體中,遠側發病較近側者預後好。
四、預後最差者為腹膜後腫瘤。
案例
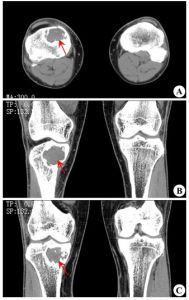 惡性纖維組織細胞瘤
惡性纖維組織細胞瘤骨的惡性纖維組織細胞瘤診治分析
一、資料與方法
1一般資料
本組52例,男33例,女19例;年齡最大75歲,最小12歲,平均年齡48.7歲。病史3個月~5年,平均病史6.8個月。本組中BMFH主要以局部疼痛、腫脹及腫塊為主訴,主要發生於膝關節和肩關節周圍(分別32例和14例,約占88%),股骨近端3例,跟骨、腰椎、髂骨各1例。術前行X線片52例,CT49例,MRI48例。X線及CT以溶骨性骨質破壞為主,常伴有軟組織包塊,MRI清楚的顯示腫瘤侵犯的範圍。
患者,女,52歲,左脛骨BMFH。術前X線片及CT示脛骨斑片樣、蟲蝕狀骨破壞,周圍由硬化邊緣(略) MRI示腫瘤侵犯的範圍、軟組織包塊及周圍比鄰的關係(略)
本組病例中術前套用取芯式穿刺活檢42例,確診為惡性纖維組織細胞瘤13例,懷疑惡性纖維組織細胞瘤18例,穿刺陽性率73.8%,未確定診斷者術中常規行冰凍快速病理。
2腫瘤分期及治療方法
本組病例有32例屬於Enneking腫瘤外科分期的ⅡB期,其中19例行保肢術,餘13例截肢。20例屬於ⅡA期,19例行保肢術,1例腰椎椎體腫瘤雖屬於ⅡA期,但因全身情況較差行保守治療,總的保肢率74.5%(38/51)。
手術方法包括:瘤段骨骨殼滅活回植內固定術、定製假體置換術、大塊瘤骨切除異體骨或自體骨移植術和截肢術等。
其他治療:術前及術後常規行化療,特殊部位(如肩胛骨腫瘤及腓骨近段腫瘤的大塊瘤骨切除)及部分ⅡB期術後行局部放療。本組中有1例髂骨腫瘤患者術前還進行了介入治療,栓塞供瘤血管,並局部注射甲胺喋呤。
保肢術在本組的早期(1995~2000年)病例中主要行瘤段骨骨殼滅活回植內固定術,並且局部套用甲胺喋啶(MTX)或者套用含MTX的骨水泥。而2000年以後,主要採用定製假體置換術。對於腓骨近段的MFH,行大段瘤骨切除而保留肢體也是一種手術方法,本組位於腓骨近段5例,其中3例行瘤骨切除,術後除行化療外,皆進行放療,餘2例行膝上截肢。本組位於肩胛BMFH4例,3例行整個肩胛骨切除,而保留上肢,1例因腫瘤侵犯臂叢及鎖骨下血管,而行肩胸截肢術。本組僅有1例骨盆腫瘤,位於髂骨翼,屬於TypeⅠ型,術前進行局部血管栓塞,行髂骨大部切除後瘤骨骨殼切除後回植,術後行化療,局部放療。
術前化療者45例,余病例因肝腎功能等全身因素的影響未能行術前化療或者未完成術前化療,所有病例皆行術後化療,術前及術後化療方案均採用順鉑(120mg/m2)加表阿黴素(60mg/m2)。對於ⅡB期保肢術的患者及部分ⅡA期的患者術後採用分割放療,劑量60~65Gy的治療。
二、結果
1隨訪結果
52例病例中36例患者進行長期隨訪,時間8~96個月,平均36.8個月,長期隨訪率69.2%(36/52)。隨訪結果發現腫瘤局部復發的9例,復發率17.3%(9/52),其中瘤骨滅活再植術後復發3例,2例再次手術行截肢,1例行定製假體置換;2例假體旁復發,骨質破壞且有軟組織包塊,均行截肢;4例軟組織復發,其中1例同時伴有肺轉移。所有復發病例加大化療劑量並局部行放療。6例患者術後2年內發生肺轉移,3例術後半年臨近淋巴結轉移,5年內全部死亡。9例術後3年內死亡。1~2年生存率100%,3年生存率75%(27/36),5年生存率61.1%(22/36)。
2保肢術後Enneking功能評分
36例長期隨訪的病例中,行保肢手術的有28例(共38例),評分:14~26分,平均21分。其中大於等於23分17例,15~22分7例,小於15分4例,其中瘤段骨滅活回植術保肢術後3例,假體置換術後1例,總優良率為86%。
三、討論
惡性纖維組織細胞瘤(malignantfibroushistocytoma,MFH)是一種起源於原始間葉細胞的肉瘤,一般由組織細胞和成纖維細胞共同組成。骨惡性纖維組織細胞瘤(malignantfibroushistocytomaofbone,簡稱BMFH)多為原發,亦可繼發於畸形性骨炎、骨梗死、多發性骨軟骨瘤等,其發病率低,在原發性骨腫瘤中約占1.9%,在惡性骨腫瘤中約占4.0%,好發於中年以上,50~60歲多見,男略多於女,比例約為3∶2。
1BMFH診斷
本病臨床表現無特異性症狀,影像學具有良、惡性多種徵象,影像學表現特徵少,在診斷上必須強調臨床、病理、影像學密切結合,主要依靠病理學診斷。
患者,男,56歲,左脛骨BMFH定製假體置換術後正側位X線片(略)
BMFH好發部位以長骨幹骺端為最多,其中在膝關節兩端的股骨與脛骨最常見,其他依次為肱骨、腓骨、脊椎、肋骨、髂骨、橈骨、肩胛骨等。病程長短不一,短至1個月,長至20年,多數在1年以內。主要以局部疼痛、腫脹及腫塊為主訴,38%~48%可發生病理性骨折,累及脊椎者可導致癱瘓。對懷疑為MFH者,術前行腫物穿刺活檢檢查有助於診斷。
本組病例中術前採用取芯式穿刺活檢術,本方法簡單、安全,適合骨性等實質性腫瘤的病理取材,成功的運用於42例患者,確診為惡性纖維組織細胞瘤為15例,懷疑惡性纖維組織細胞瘤18例,穿刺陽性率73.8%。但穿刺活檢時要注意以下幾點:(1)穿刺點的選擇主要依據手術入路並且兼顧骨質破壞的薄弱處,必要時應在C型臂X線機透視下進行。(2)儘量避開重要血管及神經進行穿刺。(3)應由有經驗的醫師進行,防止腫瘤種植轉移。
BMFH由於缺乏特殊的影像學表現,故不能根據術前X線片、CT、MRI明確診斷,但影像學檢查可以幫助診斷,並且對確定手術方案有很大幫助。根據本組資料,作者總結BMFH影像學表現:(1)X線表現:BMFH的X線表現多種多樣,特徵性少。以蟲噬狀、斑片狀或大片狀溶骨性破壞為主,邊界不清,少數可有邊緣硬化,常伴有軟組織腫塊,骨膜反應無或輕微,這是BMFH的重要特點。(2)CT表現:更清楚地顯示骨質破壞和軟組織腫塊,骨質破壞同X線,少數病灶為囊狀或膨脹性,內有粗條索狀及格線狀嵴,並見邊緣硬化,類似良性病變,骨膜反應少見。(3)MRI表現:BMFH的MRI表現缺乏特異性,但其對腫瘤髓內侵犯、周圍邊界可清楚顯示,指導手術切除範圍,還可顯示腫瘤與鄰近血管的關係以及判斷腫瘤術後改變與術後復發等方面優於X線及CT。
病理學檢查是診斷BMFH的主要依據,直接決定本病的診斷。由於BMFH的組織來源尚未明確,給病理診斷帶來了很大困難。有報導認為BMFH表現了由骨間充質細胞向纖維源性細胞表型和組織細胞表型的多種分化潛能,有一定的吞噬活性和明顯異型、多形性,容易造成誤診。但近年來,隨著免疫組織化學技術和電鏡技術的廣泛套用,使作者對BMFH有了新的認識,也給了作者新的診斷方法。如BMFH腫瘤細胞均表達vimentin,並不同程度的表達了Lysozyme、AAT、AACT、CD68和Mac387,部分表達actin、desmin,這些有助於提高對BMFH的病理學診斷。
2、BMFH的鑑別診斷
(1)溶骨性骨肉瘤:常見於青少年,病程短,疼痛明顯,骨皮質破壞嚴重,骨膜反應多而顯著,並有腫瘤新生骨,AKP升高;(2)尤文氏肉瘤:發病年齡小,臨床症狀明顯,常有發熱及白細胞升高,多見於長骨骨幹,常有篩眼樣骨質破壞,可出現骨質硬化和骨髓腔膨大,可見廣泛蔥皮樣骨膜反應;(3)骨巨細胞瘤:多見於青壯年,病變多從骨端開始,呈偏心性分房狀膨脹性骨質破壞,邊界清晰,皮質菲薄,無骨膜反應,無軟組織腫塊;(4)轉移性骨腫瘤:常在中年以後發病,有原發腫瘤病史,常多發,多見於脊椎、扁骨、股骨上段,膝關節以下很少累及,以溶骨型常見,發生於長骨者多在骨幹或鄰近的乾骺端,一般無骨膜反應和軟組織腫塊。BMFH最後確診依賴於組織病理學檢查。
3、BMFH治療
BMFH的治療關鍵在於早期發現、及時治療,要獲得理想的效果則取決於治療的正確性和徹底性。治療以手術為主,配合術前、術後化療及術後放療等綜合治療。(1)手術。手術的方法包括:四肢主要採用腫瘤切除加定製假體置換、截肢、腫瘤切除瘤骨骨殼滅活再植術、腫瘤切除自體骨移植或異體骨移植術。手術切除範圍一般可根據術前組織學和影像學診斷作出估計,主要包括皮膚、皮下組織和距腫瘤邊緣2~3cm的正常組織以及活檢或引流的傷口。本組手術切除範圍主要依據MRI,截骨平面為MRI所示腫瘤骨幹側邊界外30mm處。
手術方式的選擇:由於大多數的BMFH為高度惡性腫瘤,腫瘤分期為ⅡA和ⅡB期,保肢手術適用於ⅡA期和對化療敏感的ⅡB期腫瘤,病灶周圍大的神經血管未受累,可在安全邊界完整切除且通過重建可以獲得較好的功能。本組病例19例ⅡA期,19例ⅡB期皆進行了保肢術。保肢術中由於早期假體設計本身存在的局限性,套用較少,主要以腫瘤切除瘤骨骨殼滅活再植術、腫瘤切除自體骨移植或異體骨移植術為主。由於術後併發症主要集中在瘤段骨滅活回植術後,包括復發、骨不癒合等,但近些年隨著假體設計技術及理念的提高,套用範圍大大擴大,逐漸成為保肢術的主流。本組病例主要採用腫瘤切除加定製假體置換和腫瘤切除瘤骨骨殼滅活再植術,腫瘤切除自體骨移植或異體骨移植術採用較少,可能與植骨的來源、腫瘤累及的部位及病例數較少有關。(2)輔助放療和化療。
目前最常用的放療方式是術後針對腫瘤行外照射。常規採用分割放療,60~65Gy的中等劑量放射可以根除亞臨床病灶。而且作者發現本組16例術後放療的患者中僅有1例復發,2例轉移,也體現了BMFH的又一特性:對放療的敏感性。化療、特別是新輔助化療的套用對惡性腫瘤的保肢治療提供了可靠的保障。bacci等用輔助化療加手術切除治療MFH患者12例,存活率為59%,同期單用手術治療的18例只有1例存活,認為化療不僅能控制微小的腫瘤病灶還能降低局部復發率。本組BMFH採用ADM+DDP化療方案(ADM:表阿黴素,DDP:順鉑),這一方案在骨肉瘤的化療中起到了很好的控制復發和轉移的作用,通過本組的隨訪結果可以看出本方案對BMFH同樣有一定的作用,本組病例的5年生存率達61.1%(22/36),與Bacci等所報導的相近,但本組中仍然有很高的復發率和轉移率,作者分析可能與化療的套用時間、用藥劑量以及腫瘤本身的高度侵襲性等有關。
4、BMFH治療預後
腫瘤的組織學分級被認為是與遠處轉移和生存率相關的最重要的因素之一,腫瘤體積,位置的深淺也與其相關。而外科手術的徹底性與否,直接決定腫瘤的預後。本組隨訪病例中,發生的9例復發病例,分析原因可能與手術本身切除的徹底性及選擇的手術方式等有關。MFH以血道轉移為主,最常見的部位是肺,其次有肝、骨、腦等,但淋巴轉移也很常見。本組中6例MFH發生肺轉移,3例淋巴結轉移,皆於術後2年內發生,可能與術後不能堅持化療等有關。
