
簡介
 成骨不全症
成骨不全症 脆骨病是成骨不全症的俗稱,也被稱為“玻璃娃娃”或是“瓷娃娃”症,為先天性發育障礙。男、女發病相等,15%以上的病人有家族史。本病呈常染色體顯性或隱性遺傳方式,可為散發病例。藍鞏膜的傳遞為 100%,聽力喪失依年齡而異。散發病例多因新突變所引起,常與父母高齡有關。
成骨不全症的發生機率為萬分之一以下,被列為罕見疾病。截至2012年,中國有10萬脆骨病患者。
類型
成骨不全症有許多分類方法:根據第1次發生骨折的時間早晚,分為先天型及遲髮型;根據病情輕重分為3型;Sillence於1979年根據遺傳方式和臨床表現將其分成4種類型,這一分類套用最為廣泛。
根據病情分型
1、胎兒型:病情嚴重,常見顱骨骨化不全,胎兒期已有多次骨折,大多是死胎或生後短期夭折。
2、嬰兒型:較少見,出生後可有骨折,以後較輕微的外傷,甚至無外傷都可造成多發性骨折,女性患者多於男性,藍色鞏膜及韌帶鬆弛多見。
 成骨不全症
成骨不全症 3、少年型(遲髮型):病情最輕,出生時可以沒有骨折,兒童期容易發生骨折,到青春期後有自動改善的趨勢,20歲前後可因耳硬化造成耳聾。
診斷鑑別
一般並不困難。有時要與嚴重的佝僂病相區別。佝僂病表現為骨骺軟骨增寬、模糊、乾骺端到鈣化軟骨區不規則,分界不清。乾骺端本身呈杯狀增寬。此外,其它骨骼的稀疏情況不及成骨不全症者明顯。臨床上尚應與軟骨發育不全,先天性肌弛緩,甲狀腺功能減退及甲旁亢等區別,一般說來並不困難。病理診斷引起下背痛的原因還有很多,先天性骨缺陷,退行性疾病或骨畸形可作X線檢查,如攝取顯示椎間小關節面的斜位片。椎間盤破裂,韌帶扭傷和肌肉撕裂為突然發病,症狀常在舉重物後24小時內開始。特定部位局部壓痛和肌肉痙攣是有意義的,提示為背部本身的病變而不是骨盆內或腹膜後疾病。CT掃描或MRI檢查可提供有價值的縱軸空腔變形的圖像,骨折與骨折脫位可通過病史,創傷的性質。X線檢查,CT掃描,骨掃描(如99m鎝焦磷酸鹽標記)來排除。椎體後小關節的慢性關節炎通常與退行性椎間盤疾病有關,前者有骨關節炎的特殊臨床表現與X線徵象,後者有神經根激惹症狀。過度伸展通常加重受累椎體後小關節的疼痛,較年輕的成人逐漸發生的下背部痛提示潛在脆骨病的骨異常,如脊椎前移或脊椎關節病(如強直性脊椎炎或骶髂關節炎);青春期發病高度提示脊椎關節病,盆腔與腹膜後疾病有相應的症狀,無腰部的局部體徵。腫瘤與感染較難診斷,可類似破裂的椎間盤,占位性腫瘤常由CT,MRI或脊髓造影診斷。腦脊液檢查不一定能鑑別腫瘤與椎間盤破裂,兩者的腦脊液蛋白含量都可以升高,但此項檢查在診斷腦脊膜炎和其他感染是必要的。纖維肌痛可引起慢性下背部疼痛和僵硬感,為其局限性(肌筋膜)或瀰漫性(纖維肌痛)症狀的一部分.此外。
宜混疾病
一般並不困難。有時要與嚴重的佝僂病相區別。佝僂病表現為骨骺軟骨增寬、模糊、乾骺端到鈣化軟骨區不規則,分界不清。乾骺端本身呈杯狀增寬。此外,其它骨骼的稀疏情況不及成骨不全症者明顯。臨床上尚應與軟骨發育不全,先天性肌弛緩,甲狀腺功能減退及甲旁亢等區別,一般說來並不困難。
輔助檢查
一般均正常,有時可以有血鹼性磷酸酶的增加,這可能是由於外傷骨折後,成骨細胞活動增加所致。極嚴重者有血漿鈣及磷的減低,但極少見。
Sillence分型
根據遺傳方式及臨床表現分為:
Ⅰ型:常染色體顯性遺傳,臨床特點是骨質脆弱,生後骨折,藍鞏膜。其中又以牙齒正常為A型,成牙不全為B型。
Ⅱ型:常染色體隱性遺傳,可在圍產期死亡,存活者表現為深藍色鞏膜、股骨畸形和串珠肋。
Ⅲ型:常染色體隱性遺傳,出生時有骨折,因多次骨折骨骼畸形進行性加重,鞏膜和聽力正常。
Ⅳ型:常染色體顯性遺傳,鞏膜和聽力正常,僅表現為骨質脆弱。
病因病機
病因
具體病因不明。
發病機理
 I 型膠原纖維的合成
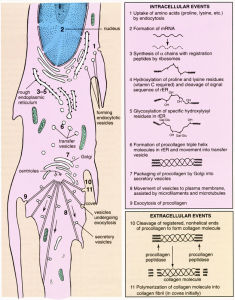
I 型膠原纖維的合成 大部分成骨不全症患兒存在編碼 I 型膠原的基因突變,導致結締組織中膠原量尤其是 I 型膠原含量下降,I 型膠原是骨骼、皮膚、肌腱、牙本質和鞏膜的主要結構蛋白,因而這些部位的病變更明顯。成骨不全症患兒的I型膠原基因突變已被廣泛研究。文獻報導編碼 I 型膠原的兩個基因有超過250種突變與成骨不全症有關。
I 型膠原為長的三螺鏇大分子,由兩條α1鏈,一條α2鏈構成,兩鏈間僅存在輕微的胺基酸序列差異。兩鏈均含有重複的胺基酸三聚體GXY:G(Glycine,氨基乙酸),X(Proline,脯氨酸),Y(hydroxyproline,羥脯氨酸)。每間隔兩個殘基為氨基乙酸殘基,這對形成螺鏇結構至關重要,因為其側鏈允許形成緊密的異構三聚體。任何一種胺基酸替代了氨基乙酸後均會破壞高度規則的螺鏇結構。
大部分 I 型成骨不全症為突變後1個等位基因靜止,因而使正常的 I 型膠原數量下降。這些突變使COL1A1基因或COL1A2基因失去功能,導致 I 型膠原合成障礙,或增加不能與正常膠原鏈結合的異常膠原鏈數量。有多種分子機制與此相關。最常見的為失效點突變,或一對/兩對插入/缺失突變,這些突變使RNA轉錄時產生提前中止編碼。另外一種可能為粘結突變,序列被切斷為不穩定的蛋白質,不能參與螺鏇形成,或轉錄在細胞內迅速降解,或轉錄後仍位於細胞核內。
II 型、III型和IV型成骨不全症患兒的I型膠原的某一鏈存在結構缺陷。其中大多數為點突變(85%),氨基乙酸被其他胺基酸殘基替代,少數情況下為粘結突變。突變的膠原鏈參與螺鏇結構形成,導致I型膠原結構改變,其臨床表現比膠原鏈完全無功能者更嚴重。大部分病例中,因為不穩定和細胞內降解往往還同時存在沉積入骨的膠原量減少。
近年來檢查I型膠原是否突變的方法有很大的進步。已經從需要皮膚活檢、纖維母細胞培養,進行RNA和蛋白分析,轉變為抽血直接進行DNA分析。這些改變提高了分子分析的速度和敏感性,為臨床診斷不典型病例、基因諮詢和產前診斷提供了新方法。
2011年前後的科學研究證實,在嚴重的成骨不全症患兒,約30%無I型膠原的結構或數量的異常。這些病例可能存在其他骨蛋白異常。
病理改變
 成骨不全症
成骨不全症 廣泛的間充質缺損,使膠原纖維成熟受抑制。在軟骨化骨過程中,骨骺軟骨及軟骨鈣化區均正常,但在乾骺端成骨細胞及骨樣組織稀少,形成的骨小纖細稀疏,呈縱向排列,無交叉的骨小梁可見。膜內化骨過程亦受影響,骨膜增厚但骨皮質菲薄,且缺管板層狀結構,哈佛氏管腔擴大,骨髓腔內有許多脂肪及纖維組織,骨較正常短,周徑變細,兩端膨大呈杵狀。顱骨甚薄,可見有分散的不規則的鈣化灶,嚴重者像一個膜袋,囟門延遲閉合。皮膚及鞏膜等亦有病變。成骨不全是基因變異的典型例證,近年來在生化、細胞超微結構和分子水平已有不少的研究。
1、骨粘連蛋白(osteonectin)、聚糖蛋白(proteog1ycan)減少,聚糖蛋白從膠原纖維分離,嚴重者聚糖蛋白顆粒與膠原的交織減少達95.2%。
2、骨組織內膠原類型改變,正常骨組織中只有 I 型膠原,II 、III、IV型少。而成骨不全的長骨有多量III型膠原。
3、骺生長板肥大,原始礦化區礦化紊亂,小柱斷裂、礦化很差的區域,葡萄糖氨基糖胺(GAGS)基質有生化改變。膠原分子缺陷、GAGS改變、非膠原蛋白改變是成骨不全的主要病理改變基礎。
4、骨膜增厚、微血管形成缺陷,動脈與毛細血管壁增厚,管腔為增生的內皮細胞與肌細胞所阻塞,骨膜細胞培養增殖率高,胞漿內磷脂、滑面內質網增加,溶酶體、粗面內質網減少。
5、灰石結晶,特別是小磷灰石結晶減少,II 型最顯著, I 型兒童期減少,青年以後有所好轉,而III型和IV型青年期也減少,特別是3型更明顯。
6、發現來自成骨不全病人的骨細胞與骨疵細胞的培養細胞對生長轉移因子(TGF-B)沒有反應,而對來自正常人骨痴細胞的培養細胞,TGF—B在刺激膠原合成的同時,增加鹼性磷酸酶的活性。說明成骨不全的骨細胞是處於不同程度的末成熟階段。
臨床表現
 成骨不全症
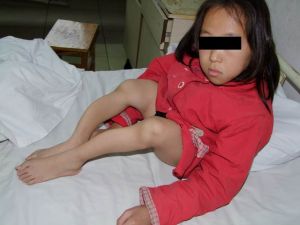
成骨不全症 1、骨脆性增加。輕微的損傷即可引起骨折,嚴重的病人表現為自發性骨折。
先天型者在出生時即有多處骨折。骨折大多為青枝型,移位少,疼痛輕,癒合快,依靠骨膜下成骨完成,因而常不被注意而造成畸形連線。長骨及肋骨為好發部位。多次骨折所造成的畸形又進一步減少了骨的長度。青春期過後,骨折趨勢逐漸減少。脆骨病,是一種先天性骨胳病。其特徵為骨質脆弱,容易骨折。
2、藍鞏膜。
約占90%以上。這是由於患者的鞏膜變為半透明,可以看到其下方的脈絡膜的顏色的緣故。鞏膜的厚度及結構並無異常,其半透明是由於膠原纖維組織的性質發生改變所致。
3、耳聾。
常到11~40歲出現,約占25%,可能因耳道硬化,附著於卵圓窗的鐙骨足板因骨性強直而固定所致,但亦有人認為是聽神經出顱底時受壓所致。
4、關節過度鬆弛尤其是腕及踝關節。
這是由於肌腱及韌帶的膠原組織發育障礙。還可以有膝外翻,平足。有時有習慣性肩脫位及橈骨頭脫位等。
5、肌肉薄弱。
6、頭面部畸形。
嚴重的顱骨發育不良者,在出生時頭顱有皮囊感。以後頭顱寬闊,頂骨及枕骨突出,兩顳球狀膨出,額骨前突,雙耳被推向下方,臉呈倒三角形。有的患者伴腦積水。
7、牙齒發育不良。
牙質不能很好的發育,乳齒及恆齒均可受累。齒呈黃色或藍灰色,易齲及早期脫落。
8、侏儒。
這是由於發育較正常稍短,加上脊柱及下肢多發性骨折畸形癒合所致。
9、皮膚疤痕寬度增加,這也是由於膠原組織有缺陷的緣故。
檢查診斷
診斷標準
2、 藍鞏膜。
3、牙質形成不全(dentinogenesis imperfecta)。
4、早熟性耳硬化(premature otoclerosis)。
上述4項中出現2項特別是前2項,即可診斷。結合 影像學檢查有助於診斷。
X線檢查
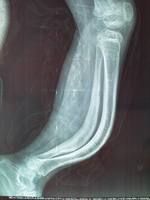
X線主要表現為骨質的缺乏及普遍性骨質稀疏。
1、在長骨表現為細長,骨小梁稀少,呈半透光狀,皮質菲薄如鉛筆畫。髓腔相對變大,嚴重時可有囊性變。骨兩端膨大呈杵狀,可見有多處陳舊性或新鮮骨折。有的已經畸形連線,骨幹彎曲。有一些畸形是因肌肉附著處牽拉所致,如髖內翻、股骨及脛骨呈弓形。某些病人在骨折後會形成豐富的球狀骨痂,其數量之多,範圍之廣,使人會誤診其為骨肉瘤。另有一些病人的骨皮質較厚,稱“厚骨型”。少見。
2、顱骨鈣化延遲,骨板變薄,雙顳骨隆起,前囪寬大,岩骨相對緻密,顱底扁平。乳齒鈣化不佳,恆齒髮育尚可。
3、椎體變薄,呈雙凹形,骨小梁稀少,椎間盤呈雙凸形代償性膨大。可以有脊柱側彎或後突畸形。
4、肋骨從肋角處向下彎曲,常可見多處骨折。骨盆呈三角形,盆腔變小。
輔助檢查
一般均正常,有時可以有血鹼性磷酸酶的增加,這可能是由於外傷骨折後,成骨細胞活動增加所致。極嚴重者有血漿鈣及磷的減低,但極少見。
鑑別診斷
1、佝僂病:有時要與嚴重的佝僂病相區別。
佝僂病表現為骨骺軟骨增寬、模糊、乾骺端到鈣化軟骨區不規則,分界不清。乾骺端本身呈杯狀增寬。此外,其它骨骼的稀疏情況不及成骨不全症者明顯。
2、臨床上尚應與軟骨發育不全,先天性肌弛緩,甲狀腺功能減退及甲狀旁腺機能亢進等區別,一般說來並不困難。
治療措施
臨床上用於治療該症的雙磷酸鹽類藥物雖在醫保目錄里,但該藥的適應症里並未列明成骨不全症,因此患者需自費。
1、無特殊治療。主要是預防骨折,要嚴格的保護患兒,一直到骨折趨減少為止,但又要防止長期臥床的併發症。
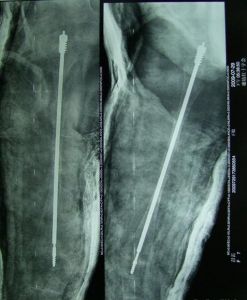
2、對骨折的治療同正常人。但骨折癒合迅速,固定期可短。
3、在矯正畸形方面,以防止再骨折為主。如皮質太薄,手術有困難時,可用異體骨移植。
4、對失聽患者,可做鐙骨切除。
5、50%~70%的病兒有脊柱側突畸形,可用支架保護。若脊柱側彎超過60°時,應矯正後作脊柱融合術。
7、文獻上有人試用降鈣治療本病,但療效不肯定。
法律保障
美國:1983年頒布了《罕見病用藥法》 ,對罕見病臨床研究費用減免稅金,並提供研究資助,罕見病患者享有政府醫療保健計畫和商業保險雙重保障。
中國台灣:2000年頒布《罕見病防治及藥物法》,患者使用的藥物及維持生命所需的特殊營養品費用施行全額報銷。
盤點常見的遺傳病
| 隨著分子生物學的發展,科學家們成功的把基因和疾病聯繫在一起。人們可以預先通過基因篩查來預測可能罹患的遺傳病。比如,GOOGLE公司創始人之一謝爾蓋?布林,就在其妻子的23andme生物技術公司通過測序發現自己可能在未來患帕金森氏綜合症,他積極通過各種方式來預防疾病的發生並投入巨資進行相關的研究。但如同一把雙刃劍,基因篩查也使一些攜帶突變基因的人在社會各個方面受到歧視,讓我們來關注一下遺傳疾病吧! |
| 黑棘皮症 | 短趾症 | 家族性高膽固醇血症 | 白化病 | 苯丙酮尿症 | 紅綠色盲 | 抗維生素D佝僂病 | 先天性髖關節脫位 | 脊柱裂 | 先天性聾啞 | 蠶豆病 | 強直性肌營養不良 | 亨廷頓氏病 | 老年性痴呆症 | 精神分裂症 | 脆性X綜合徵 | 原發性高血壓 | WILSON 氏病 | 成人多囊腎病 | 馬凡綜合徵 | 侏儒綜合徵 | 視網膜色素變性 | 糖原貯積症 | 神經鞘脂貯積症 | 黏多糖貯積症 | 半乳糖血症 | 白癜風 | 性反轉綜合徵 | WILMS 瘤 | 視網膜母細胞瘤 | 成骨不全病 | 自毀容貌症 | 家族性多發性結腸息肉病 | 強直性脊柱炎 | 假肥大性肌營養不良 | 牛皮癬 | 多囊腎 | 脆骨病 | 神經纖維瘤 | 上瞼下垂 | 類風濕性關節炎 | 癲癇 | 先天性心臟病 | 高膽固醇血症 | 唇裂 | 齶裂 | 魚鱗症 | 多指 | 著色性乾皮病 | 腓肌萎縮症 | 並指 | 畸形足 | 青光眼 | 全身自化 | 結腸息肉症 | 先天聾啞 | 原發性小睪症 |
