概述
 異染性腦白質營養不良異染性腦白質營養不良(metachromaticleukoencephalopathyMLD),亦稱異染色性白質萎縮或異染色性白質腦病1910年由Alzheimer首先報導系常染色體隱性遺傳性疾病,由芳基硫脂酶A(arylsulfatase-A)缺乏所引起。據國內史玉泉呂傳真等報導,本病發病率為1/13萬~1/4萬。
異染性腦白質營養不良異染性腦白質營養不良(metachromaticleukoencephalopathyMLD),亦稱異染色性白質萎縮或異染色性白質腦病1910年由Alzheimer首先報導系常染色體隱性遺傳性疾病,由芳基硫脂酶A(arylsulfatase-A)缺乏所引起。據國內史玉泉呂傳真等報導,本病發病率為1/13萬~1/4萬。病兒於生後1~2歲半逐漸出現步行困難伴四肢無力,共濟失調或肢體強直,有進行性痴呆視神經萎縮、深腱反射消失、神經傳導時間延長及腦脊液蛋白增高等。少數於3~10歲發病(少年型)或成人期發病(成人型)診斷靠白細胞硫酸酯酶A活性減少<40nmol/(h·ml)或基因診斷。
病因
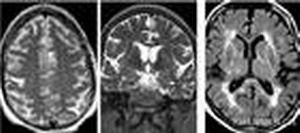 異染色性白質腦病異染色性腦白質營養不良屬常染色體隱性遺傳,突變基因位於第22號染色體上。由芳基硫脂酶A(arylsulfatase-A)缺乏所引起。異染性腦白質營養不良(metachromaticleukodystrophy,MLD)又稱為腦硫脂沉積病(sulfatidosis),常染色體隱性遺傳,是芳基硫酸脂酶A缺陷所致的髓鞘形成不良。由於編碼溶酶體芳基硫酸脂酶A(arylsulfataseA,ASA)的基因MLD突變所引致,MLD位於22q13.33,其突變種類較多;大致可分為兩組:I型突變的患者不能產生具有活力的ASA,其培養細胞中無ASA活性可測得;A型突變患者則可合成少量具有活力的ASA。患者的表型取決於其基因突變的種類:I型突變的純合子或具2個不同I型突變者在臨床上表現為晚期嬰兒型;具有I型和A型突變各一者為青、少年型;而2個突變均為A型時,則呈現為成年型。少數本病患者,特別是青少年型的發病不是由於MLD突變所致,其ASA活力正常,這是由於患者缺少一種溶酶體蛋白,硫酸腦苷酯激活因子(SAP1)所造成的。這類患者亦稱為"激活因子缺乏性異染性腦白質營養不良"。
異染色性白質腦病異染色性腦白質營養不良屬常染色體隱性遺傳,突變基因位於第22號染色體上。由芳基硫脂酶A(arylsulfatase-A)缺乏所引起。異染性腦白質營養不良(metachromaticleukodystrophy,MLD)又稱為腦硫脂沉積病(sulfatidosis),常染色體隱性遺傳,是芳基硫酸脂酶A缺陷所致的髓鞘形成不良。由於編碼溶酶體芳基硫酸脂酶A(arylsulfataseA,ASA)的基因MLD突變所引致,MLD位於22q13.33,其突變種類較多;大致可分為兩組:I型突變的患者不能產生具有活力的ASA,其培養細胞中無ASA活性可測得;A型突變患者則可合成少量具有活力的ASA。患者的表型取決於其基因突變的種類:I型突變的純合子或具2個不同I型突變者在臨床上表現為晚期嬰兒型;具有I型和A型突變各一者為青、少年型;而2個突變均為A型時,則呈現為成年型。少數本病患者,特別是青少年型的發病不是由於MLD突變所致,其ASA活力正常,這是由於患者缺少一種溶酶體蛋白,硫酸腦苷酯激活因子(SAP1)所造成的。這類患者亦稱為"激活因子缺乏性異染性腦白質營養不良"。發病機制
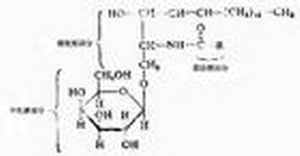 發病機制原理硫酸腦苷脂分布於神經組織髓鞘腎小管上皮細胞等細胞膜中正常情況下,芳基硫酸脂酶A催化硫酸腦苷脂水解,將半乳糖硫酸腦苷脂分解為半乳糖腦苷脂和硫酸。此酶缺乏時引起硫酸腦苷脂於體內沉積主要的病理改變為中樞神經系統髓鞘脫失,周圍神經受累輕微。病理切片中,以甲苯染色時可見神經細胞神經膠質細胞和巨噬細胞中有紅黃色的異染物質沉積肝、腎組織亦可同時受累。
發病機制原理硫酸腦苷脂分布於神經組織髓鞘腎小管上皮細胞等細胞膜中正常情況下,芳基硫酸脂酶A催化硫酸腦苷脂水解,將半乳糖硫酸腦苷脂分解為半乳糖腦苷脂和硫酸。此酶缺乏時引起硫酸腦苷脂於體內沉積主要的病理改變為中樞神經系統髓鞘脫失,周圍神經受累輕微。病理切片中,以甲苯染色時可見神經細胞神經膠質細胞和巨噬細胞中有紅黃色的異染物質沉積肝、腎組織亦可同時受累。疾病分類
 腦白質營養不良患者腦白質內可發生許多疾病。而腦白質對各種有害刺激的典型反應是脫髓鞘變化,它可以是神經系統疾病如感染、中毒、退行性變、外傷後、梗塞缺乏等的繼發表現。有一組至今原因尚不甚明確的中樞神經系統脫髓鞘性疾病。因此,目前對這一組疾病的分類,按它們起病時賄鞘發育是否成熟,可將這組疾病分為兩個大類:①髓鞘發育正常的脫髓鞘性疾病如多發性硬化、進行性多灶性腦白質病、急性散發性腦脊髓炎等;②髓鞘形成不良性疾病如類球狀細胞型腦白質營養不良、異染性腦白質營養不良、海綿狀腦病等。
腦白質營養不良患者腦白質內可發生許多疾病。而腦白質對各種有害刺激的典型反應是脫髓鞘變化,它可以是神經系統疾病如感染、中毒、退行性變、外傷後、梗塞缺乏等的繼發表現。有一組至今原因尚不甚明確的中樞神經系統脫髓鞘性疾病。因此,目前對這一組疾病的分類,按它們起病時賄鞘發育是否成熟,可將這組疾病分為兩個大類:①髓鞘發育正常的脫髓鞘性疾病如多發性硬化、進行性多灶性腦白質病、急性散發性腦脊髓炎等;②髓鞘形成不良性疾病如類球狀細胞型腦白質營養不良、異染性腦白質營養不良、海綿狀腦病等。臨床表現
 患兒根據起病年齡可以區分為晚期嬰兒型(1~2歲起病)、少年型(4~15歲起病)和成年型(16歲以後起病),以晚期嬰兒型最為常見。
患兒根據起病年齡可以區分為晚期嬰兒型(1~2歲起病)、少年型(4~15歲起病)和成年型(16歲以後起病),以晚期嬰兒型最為常見。典型者其病程可分為以下3期:
第1期:1~2歲之間發病病前嬰兒發育正常;起病後病兒逐步出現運動減少肌張力降低步態蹣跚,維持姿勢困難不能獨立站坐,甚至抬頭困難。體格檢查可見肌張力降低,腱反射降低或消失,視盤蒼白,錐體束征陰性。腦電圖正常或有慢波增多腦脊液壓力正常,可有輕度蛋白質增高。此期持續數周至數個月
第2期:病兒進行性智慧型減退語言減少到消失對周圍環境逐步反應減少。尖叫而臥床不起體格檢查可見瞳孔光反應遲鈍視盤蒼白萎縮,面無表情,吞咽動作緩慢四肢肌張力增高肢體伸直腱反射亢進,病理錐體束征陽性,但軀幹和
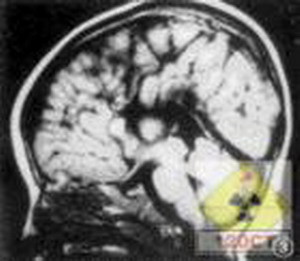 異染性腦白質萎縮
異染性腦白質萎縮頸肌肌張力正常或偏低。腦脊液壓力、細胞正常蛋白質明顯升高。腦電圖出現瀰漫性慢波灶。此期病程可持續一至數年。
第3期:為晚期病症病兒對外界極少反應常有抽搐和肌陣攣發作呈現特殊的去皮質強直體位頭後仰項強直肌強直,四肢腱反射極難引出,兩側錐體束征陽性。瞳孔散大而對光反應極差眼球遊動或是有“玩偶”眼征吸吮和吞咽嚴重障礙。腦電圖出現瀰漫性慢波和散在的棘波。腦脊液蛋白質進一步增高達1g/L以上多數病兒多次繼發感染,常於5~6歲左右病故。
少年型和成年型病者起病晚進展緩慢常有周圍神經感覺缺失晚期可有精神和行為異常。
併發症
隨病情發展,出現的症狀體徵可以是本病表現也可以看作本病不同類型的併發症。
診斷
 診斷參數本病臨床症狀與Krabbe病沒有什麼區別診斷十分困難特別是成年型患者診斷更為困難尿、血液白細胞中芳基硫酯酶A活性降低為診斷本病的依據病人皮膚成纖維細胞培養芳基硫酯酶A活性降低更為敏感。周圍神經活檢、直腸黏膜活組織檢查中發現異染色性類脂質顆粒可確診本病。本病的確診依據是ASA活力檢測,但在少數有典型症狀而ASA活力正常情況時,則應考慮激活因子缺乏性異染性腦白質營養不良的可能性。需與Pick病Alzheimer病等鑑別。
診斷參數本病臨床症狀與Krabbe病沒有什麼區別診斷十分困難特別是成年型患者診斷更為困難尿、血液白細胞中芳基硫酯酶A活性降低為診斷本病的依據病人皮膚成纖維細胞培養芳基硫酯酶A活性降低更為敏感。周圍神經活檢、直腸黏膜活組織檢查中發現異染色性類脂質顆粒可確診本病。本病的確診依據是ASA活力檢測,但在少數有典型症狀而ASA活力正常情況時,則應考慮激活因子缺乏性異染性腦白質營養不良的可能性。需與Pick病Alzheimer病等鑑別。檢查
 尿血液白細胞檢查實驗室檢查
尿血液白細胞檢查實驗室檢查
尿血液白細胞中芳基硫酯酶A活性降低。其它輔助檢查
1.周圍神經活檢、直腸黏膜活組織檢查中發現異染色性類脂質顆粒。
2.CT檢查見基底節和小腦有鈣化,片狀脫髓鞘。有末梢神經病。
治療
 懷孕期測定羊水細胞本病患者在症狀尚未出現以前可考慮進行骨髓移植,以延緩或終止病情發展;對神經系統已有廣泛病變者尚無滿意治療方法。
懷孕期測定羊水細胞本病患者在症狀尚未出現以前可考慮進行骨髓移植,以延緩或終止病情發展;對神經系統已有廣泛病變者尚無滿意治療方法。曾有人套用牛腦提取的芳基硫脂酶A1000萬U靜脈或鞘內注射雖然在治療以後肝臟組織中酶的活性恢復正常但腦內酶活性和脫髓鞘病變仍無任何改善。本病主要是對症治療。用從人尿中提取的芳基硫酸酯酶A進行治療,取得了一定的療效。由於本病對小兒危害較大,死亡率高,且為遺傳性疾病,所以我們應對有此家族史的下一代在母親懷孕期就測羊水細胞內芳基硫酸酯酶A的活性,確診後應終止妊娠。
預防
 近親結婚的危害多數患兒多次繼發感染而於5~6歲病故少年型和成年型患者進展緩慢,常有周圍神經感覺缺失。晚期可有精神和行為異常,可進行遺傳諮詢。預防措施包括避免近親結婚、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生。
近親結婚的危害多數患兒多次繼發感染而於5~6歲病故少年型和成年型患者進展緩慢,常有周圍神經感覺缺失。晚期可有精神和行為異常,可進行遺傳諮詢。預防措施包括避免近親結婚、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生。相關詞條
