
概況
 亨廷頓病性痴呆
亨廷頓病性痴呆亨廷頓病以常染色體顯性並完全外顯形式遺傳。即一個患者的後代將有半數發病。男性和女性的發病可能性相同。平均起病年齡35~40歲,也可早至2.5歲,晚至75歲。平均病程14年。
亨廷頓病的流行病學調查已有多篇報導。由於調查方法及對象不同,結果差異較大。亨廷頓病的患病率從0.5/10萬人口至7.8/10萬人口,日本報導本病的患病率為0.11/10萬人口。而委內瑞拉在圍繞Maraeaibo湖地區,亨廷頓病的患病率最高,達到700/10萬人口。國內尚無亨廷頓病的專題流行病學調查,但臨床並非罕見。至今已報導100餘家系,分布於全固各地區。
病因
 亨廷頓病性痴呆遺傳研究
亨廷頓病性痴呆遺傳研究已證明亨廷頓病為常染色體顯性並完全外顯形式遺傳。父或母患病,子女的發病機會是50%。本病好發於阿利安族的白種人。Huntington所報告的病例都是居住美國的英國移民後代,大約1000多例患者其共同的祖先是17世紀從英國移民美國的兩兄弟。我國李氏等(1980)首先報導國內亨廷頓病的家系調查。事實上本病可見於世界各地。我國至今已報告亨廷頓病100多個家系。Singer發現香港一個中國人的家系四代24個成員中有7例患病。
亨廷頓病的群體基因頻率為10.69×10~,攜帶者的頻率為0.5/萬~1/萬,突變率為0.7×10一6。父系遺傳占優勢者發病較早,而母系遺傳占優勢者發病較晚,但如母親患病在妊娠過程中由於母體與胎兒的相互作用,大部分胎兒流產。而由父親患病,小孩多能存活,但以後可能發病。因此認為有限制遺傳傾向。李氏報導22個家系中,7例系母系遺傳,15例系父系遺傳。在高氏報告追蹤觀察的家系中,第四代已確診亨廷頓病者7例,僅2例是母親遺傳,5例為父親遺傳。本病不隔代遺傳,如不攜帶有病的基因則不發病。如發現有隔代遺傳現象,可能因觀察時間不夠。高氏觀察的亨廷頓病家系中Ⅸ,已發病,但其父Ⅲ2除脾氣變得暴躁外。神經檢查正常,追蹤觀察的第8年用WAIS查智慧型發現其智商95(低於同文化水平的對照組),第9年才出現不自主運動,比其子患亨廷頓病肯定診斷晚9年;實際上。其性格改變應認為已發病。亨廷頓病的遺傳大多呈現早發傾向。且一代比一代重。
分子生物學的研究,發現亨廷頓病基因位於第4號染色體短臂上(4P16.3)/20J。通過限制性片段長度多態性(RFLP)連鎖分析,為亨廷頓病的基因診斷提供了可能。1993年終於發現了亨廷頓病基因,並且揭示了ITl5基因的不穩定突變。金氏等套用G8亞克隆探針對家系亨廷頓病中部分成員進行RFLP及單體型分析,結果表明中國人D4SIO區域可能有自己的特點。與國外報導不一致。曾氏等套用RFLP連鎖分析和巢式聚合酶鏈反應直接檢測ITl5基因CAG三核苷酸重複序列的技術,對兩大亨廷頓病家系中8例亨廷頓病患者和39例高風險家庭成員做基因診斷。結果表明39例亨廷頓病高風險者的症狀前預測的結果是有名為亨廷頓病基因攜帶者。結果也表明ITl5基因的不穩定性突變是導致中國人亨廷頓病的遺傳學基礎。
興奮性中毒性假說
已證明興奮性胺基酸具有神經毒性,亨廷頓病雖為遺傳缺陷造成,但其病理上則以紋狀體的選擇性細胞喪失為特點。提示可能與興奮性毒物有關。在給小鼠紋狀體內注射了Kainate後,可使原有的γ一氨基丁酸和膽鹼能標記物丟失而誘發本病。但由此造成的動物槨型其細胞損害缺乏應有的選擇性。喹啉酸(QuinolinicAcid)是谷氨酸的中間代謝產物,目前被認為是最有可能導致此病的內源性毒物。在實驗小鼠紋狀體內注射喹啉酸後不僅可出現興奮性神經元損害的病理改變,而且還可見到選擇性細胞損害,這些變化同人類患此病時的病理改變一致。由於尚未發現亨廷頓病時喹啉酸濃度升高現象,故可以初步推論亨廷頓病的病人中可能先已存在某種代謝的異常,使某些細胞對正常濃度的谷氨酸、喹啉酸或其它內源性胺基酸的敏感性增高,並由此造成了細胞的變性死亡。為此提出一種假說,亨廷頓病基因可能通過改變谷氨酸傳遞系統產生影響。因谷氨酸是一種豐富的興奮性神經遞子系統,而且含有豐富谷氨酸纖維,從皮層進入紋狀體。這樣假說仍是一種推測,但提供一種有意義的可能病因。
氧化磷酸化假說
Machlin等(1987)和Choi(1988)觀察到在神經培養中過度刺激N-甲基D-天門冬氨酸(NMDA)受體,產生鈣過度進入神經細胞內導致自由基(過氧化物)對神經元毒性。Miyamoto等(1988)報告用抗氧化左鏇生育酚和idebenone能夠預防這樣損傷,因而考慮到氧化磷酸化假說。
來自父親遺傳的患者發病年齡較早。提出線粒體基因可能決定發病年齡的可能性。在氧化磷酸化中所需酶的產生是由線粒體基因控制的。對細胞中氧化酶系統作用愈強,細胞中有毒的自由基濃度愈低。幾乎所有提供給胎兒的線粒體來自母親卵子的細胞漿,而精子在其有限的細胞漿中含量相當少。
由具有亨廷頓病基因的婦女生育的兒童,其發病年齡肯定很遲。這類人有很強的氧化磷酸化系統遺傳到她的後代。患有亨廷頓病的男人。作為父親的氧化磷酸化系統的效力有選擇性。但由無病的母親遺傳的線粒體則反應一般人氧化磷酸化效力。
具有亨廷頓病基因的人,結合具有有效的氧化磷酸化系統,發病時間傾向延遲。因為自由基不具有效活性,因而減慢了神經元的死亡速度。反之具有雙重無效力的氧化磷酸化系統者,亨廷頓病基因由於不失活致更早發病。
症狀
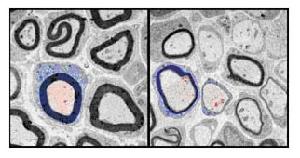 亨廷頓病性痴呆細胞分析
亨廷頓病性痴呆細胞分析運動障礙
亨廷頓病患者的運動障礙包括不隨意和隨意運動障礙兩方面。不隨意運動障礙表現為舞蹈症、肌張力不全、手足徐動和運動不安。舞蹈樣不自主運動是本病最突出的特徵。大多開始表現為短暫的裝鬼臉、點頭和手指屈伸運動。運動類似抽搐,但較慢且非刻板式。隨著病情發展,症狀逐漸進展,運動的不隨意性質愈益明顯。不隨意運動累及面、頸、肢體和軀幹。嘴一張一合,抬眉毛和頭屈曲均為典型動作。當患者行走時,軀幹和腿呈不規則的一瘸一拐,蹣跚和皺行,加上手變換著伸鏇前和屈鏇後的姿勢,步態表現像舞蹈。嚴重者因全身不自主運動而不能站立和行走。即使坐著也不穩,身體扭動,突然站起又突然坐下。
病情發展至只能臥床,全身不停的扭動,甩胳膊、蹬腿。雖然在病的早期,以不隨意運動障礙為主要特徵,當病情發展時,隨意運動受損愈趨明顯。表現為動作笨拙,遲緩,僵直,不能維持複雜的隨意運動,以致吞咽困難,構音障礙。有些患者表現不正常的眼運動,包括隨意注視的核上性障礙,視動眼震受員和快速眼一運動睡眠的眼快速活動受損。在病的晚期,運動速度減慢而具有手足徐動症和肌張力不全的特徵。最後患者呈固定的四肢癱姿勢,隨意運動消失。肌張力多為正常,典型病例直到晚期肌張力才減退。部分患者腱反射增高。感覺不受累。少數患者可伴有癲癇,肌陣攣。
雖然舞蹈樣運動障礙是亨廷頓病的典型運動障礙,但少數病例表現為不動性肌強直的變異型(Westphal變異型),與帕金森病相似。12%~14%的亨廷頓病患者以肌強直為主要症狀。在少年起病的亨廷頓病患者中肌強直特別常見。少年型(20歲以前起病者)占所有享廷頓病的5%~10%。左氏等統計國內已報導的亨廷頓病病例中。少年型占7%。約1/3~1/2少年型亨廷頓病的主要運動異常為肌強直而非舞蹈樣動作。運動異常表現為肌強直、肌陣攣,至晚期甚至呈角弓反張。與成人相反,少年型亨廷頓病的癲癇發病率增高,表現為全身性發作者可多達50%。
認知障礙
痴呆是亨廷頓病患者恆有的特徵之一。MeHugh和Folstein於1975年首先提出亨廷頓病患者的認知障礙具有皮質下痴呆的特徵,即記憶缺陷、認知緩慢、淡漠和抑鬱。與皮質性痴呆如阿爾茨海默病不同的是無失語、失用和失認。
認知缺陷在亨廷頓病的早期即可出現。開始輕的缺陷表現在日常生活和工作的記憶和計算、詞的流利性、視空間功能及對社會和人際關係的判斷、日常生活的速度能力和結構判斷上。從一個任務到另一個任務同時探索幾件事的變換上也可表現患者的認知障礙先兆。
記憶障礙常於早期發生。記憶損害的模式與阿爾茨海默病早期或柯爾薩柯夫綜合徵時所見遺忘不同。後兩種病的患者是記住新信息能力首先破壞,而對老信息的回憶相對保留。亨廷頓病患者對記住近期材料和回憶遠期老的信息同樣困難。仔細分析記憶缺陷發現最初記住新信息僅有輕度損害,而將信息作修飾以便有效儲存則有明顯困難,回憶有顯著缺陷。
口語改變包括完成口語流利性測驗不良(一分鐘內儘可能多地說出一類東西的名稱),輕度找詞困難和構音障礙。口語流利性損害是亨廷頓病最早能計量查出的認知功能不正常之一,比記憶和其他認知功能衰退出現得早。在病的中期和晚期,患者不能完成需要組織,連續和語言學精心加工的語言測驗,也不能完成需回憶不常用詞的命名測試。但這些測驗超出語言範圍,因還需要記憶和認知能力。沒有皮質病變時典型的錯語症錯誤和失語症的其他語言異常,但構音和韻律障礙為患者的突出特徵。舞蹈樣運動障礙常累及舌和唇,破壞了發音的韻律和敏捷性。橫膈運動妨礙了說話的量、速度、節律和短語的長度,使口語呈一種暴發性性質。雖然患者有構音和韻律障礙,但仍保留詞的識別記憶及對字的識別和對物的命名能力。因此。亨廷頓病患者能繼續與人交流。
隨病情發展,其他認知障礙逐漸明顯。集中力和判斷力進行性受損。患者缺乏啟動解決問題的行為。在需要組織、計畫和連續安排信息的任務上,患者感到特別困難,面對信息過多而不知所措。視空間能力下降。表現在認知測試中視空間功能測試極其困難。在需要連續安排運動的額葉系統測驗上(如手連續運動)有困難,但不是因患者有運動障礙所致。
正規的心理學測試發現在韋氏成人智力量表(WeehslerAdultIntelligenceScale,WAIS)中。算術、數字元號、數字廣度和圖畫排列的亞測試上困難最大。從亨廷頓病患者的WAIS的總成績上。操作智商明顯低於語言智商。這正反映出亨廷頓病患者視空間功能和需連續安排信息上,困難較大。而詞語的識記則相對保留。戴氏等對亨廷頓病患者的WAIS測試結果。表現為患者在語言測試各亞項中,知識和辭彙均能保持中等量表分,領悟和相似多可獲得中等偏低量表分,數字廣度量表分較低或中度偏低,算術最差,量表分均在低檔。操作測試各亞項的量表分大多在低檔,有些量表分雖到達中等也偏低,其中數字元號、圖片排列和圖形拼湊最差。是因為這些亞項不僅要求視空間能力,還需要連續安排信息,組成總的概念。
雖然有些作者發現有亨廷頓病風險因素者用WAIS測試,以後發病組與不發病組間測試結果有差異。認為WAIS不能作為症狀前的有用工具。但高氏等在追蹤一個亨廷頓病家系中,認為有亨廷頓病風險因素、並且WAIS測查智商偏低者,可預示該例有發病的危險引。
由於亨廷頓病的認知障礙為隱襲起病,其記憶障礙不像阿爾茨海默病的遺忘易被發覺,且其詞語的識別能力及語言交流能力相對保留,比阿爾茨海默病更不易被發現;而患者的運動異I常即使很輕也易引人注意,常認為亨廷頓病的首發症狀是運動異常。早已有作者指出,認知障查,詢問有關精神症狀,進行高級智慧型評價;做詳細的神經系統檢查;通過各種途徑確定痴呆時期。結果發現以舞蹈樣運動障礙為首發者15例,以痴呆為首發症狀者25例,10例患者兩種症狀幾乎同時發生。痴呆發生的年齡比舞蹈樣運動異常略早,但差異不顯著。高氏等對一個亨廷頓病家族追蹤,除已發病者WAIS測查結果顯示智商明顯低下外。有患病風險的親屬與年齡和文化相匹配的對照組比。智商有顯著低下。其中3例智商低於95者,1~2年後出現了舞蹈樣異常運動。已明確診斷亨廷頓病者中,至少2例是智慧型首先減退。1例10歲患者因學習迅速下降1年被迫退學,5年後才出現肌強直及癲癇發作。另一例於51歲時以一年來面手不自主多動而就診。仔細了解是該患者原為能力很強的車間主任,因工作能力下降於50歲時被停止車間主任職務。家屬認為是因生氣(心情不舒暢)致手哆嗦。高氏等認為在追蹤該家系的亨廷頓病患者中,大多以智慧型減退為首發症狀。
 亨廷頓病性痴呆
亨廷頓病性痴呆Huntington在最初描述本病症狀時就已提出“隨著本病的發展,精神受到不同程度損害。有些表現就是精神病;而另一些則精神和身體逐漸衰退直至死亡解除其痛苦"L21。他還指出這些患者所患精神病的類型常導致自殺。以後的報導均充分證實了Huntington的觀察。患者首先出現的精神狀態變化為人格改變,包括暴躁、不整潔和失去興趣。Folstein以DSM一Ⅲ的標準評定亨廷頓病患者的精神症狀。發現有情感障礙者占33%。有間歇性暴發行為者30%,4.8%有精神抑鬱障礙。15.6%酗酒。抗社會人格和精神分裂症者各占5.9%;而30%的患者無任何精神病性障礙。Folstein認為情感障礙是最多見的精神病性症狀,且多出現在運動障礙發生之前。在亨廷頓病患者中,抑鬱症狀的發生率與自殺率高是一致的。在本病男性死亡者中,自殺占7.8%,女性者占6.4%。還常有自殺未遂者。本病情感障礙發生率高。但不是反應性障礙。因為情感障礙不僅有抑鬱。且有躁狂;而且情感障礙可出現在患者的運動障礙出現之前,或了解其病及本病的家族特點之前。如對亨廷頓病的重度抑鬱障礙未能早期明確診斷或不適當的治療致不被發現,將導致自殺率高。如早期發現患者的抑鬱症狀,套用抗抑鬱劑或電休克治療可能預防自殺。已報導亨廷頓病患者自殺率有較大差別(1.6%~12.7%),反映了對患者情感障礙治療結果的不同。
亨廷頓病患者的激動性是家庭成員最難以忍受的人格改變。這種激動性可診斷為發作性暴力行為障礙,也可作為抑鬱症的複合綜合徵的一部分。Burns等提出在與阿爾茨海默病比較後,激動性、攻擊行為和情感淡漠並不能預示亨廷頓病的發病。但發現激動性與患者病前的壞脾氣呈正相關。與好脾氣則呈負相關。
隨亨廷頓病患者的精神和神經障礙進行性衰退,至最後階段,患者緘默和呆傻。死於全身衰竭或繼發感染。
檢查
 亨廷頓病性痴呆
亨廷頓病性痴呆頭部CT和MRI可證明尾狀核萎縮,其表現為突入側腦室額角外側面的膨起消失。大致觀察掃描圖形就可發現此種萎縮,但也可以在測定額角與尾狀核間之比,或雙尾狀核指數(在尾狀核頭部水平雙側腦室寬度與同一水平顱骨外側骨板間距離之比)而確定。額角與尾狀核間比小於2,或雙尾狀核指數小於1.8均符合亨廷頓病的尾狀核萎縮。
Hasselbalch等用SPECT檢查18例亨廷頓病患者和19例年齡、性別相匹配的對照組的腦血流。發現亨廷頓病患者在尾核和豆狀核有明顯的血流下降,分別下降20%和8%。大腦皮質。尤其額葉和頂葉腦血流下降11%~13%。而丘腦、內側顳葉和枕葉的腦血流在兩組間無差異。在比較認知功能測驗與區域腦血流關係後,發現圖片排列測驗和Wisconsin卡片分類測驗結果與尾核血流呈線性關係,其他認知測試則與皮質區血流有關。結果表明,亨廷頓病的皮質和皮質下結構血流減少,且與患者認知功能有關。
正電子發射斷層照像(PET)表現尾狀核區葡萄糖代謝明顯降低,而皮質代謝通常仍正常。在X線掃描發現尾狀核萎縮前,PET已顯示該區代謝活性降低。
神經病理和病理生理學
神經病理:大體上,亨廷頓病患者的腦有輕度或中度非特異性萎縮,主要累及額和頂區。冠狀切面則發現有極突出的改變。尾狀核頭部的實質結構縮小成2~3ram厚的薄帶狀,以致測腦室的額角外側壁呈突起狀。殼核也縮小,蒼白球有皺縮但較輕。
組織學改變極突出。主要是尾狀核內小聯絡神經元(高爾基Ⅱ型細胞)喪失達70%,致大神經元與小神經元的比例增高。星形膠質細胞也有喪失,但與神經細胞比,膠質細胞仍是增多的。伴有運動遲緩的患者(Westphal變異型)顯示小神經元有更廣泛的喪失。膠質指數(glialindex。每個神經細胞的膠質細胞數)增多到正常的169%,表明亨廷頓病患者腦的紋狀體中所見到的神經膠質增生是相對的而非絕對的現象。
大腦皮質3~5層也有小神經元喪失和星形膠質細胞增生,伴血管周間隙脂褐質和充滿脂質的巨噬細胞增多。有學者強調,除紋狀體外,病變也累及到大腦皮質、白質、大腦半球纖維系統。聯合纖維系統和間腦核。
在丘腦,尤其腹外側核也常有小神經元喪失,並且下丘腦也常顯示神經元喪失,特別是外側和中間部分。黑質網狀結構可見神經元喪失。雖然小腦可不受累,但在少年患者可發生蒲肯野細胞和齒狀核細胞喪失。橄欖體、腦幹薄束核和楔束核有神經元喪失,並伴有脊髓後柱神經膠質增生。前和側柱變蒼白。Kremer等證明在丘腦下部的側管核中有神經元喪失和膠質細胞增生。腦的這種多神經病理變化可以解釋亨廷頓病患者的行為和情感改變。
已證明亨廷頓病有神經遞質改變,且這些遞質改變與患者臨床症狀有關。在紋狀體、蒼白球和黑質中。抑制性神經遞質7一氨基丁酸(GABA)和對GABA起作用的酶(谷氨酸脫羧酶。GAD)減少了70%~90%。大部分GAD位於以釋放GABA為遞質的小的高爾基Ⅱ型細胞內。膽鹼乙醯化酶活性降低,導致乙醯膽鹼減少。在正常情況下。多巴胺與GABA和乙醯膽鹼成動力平衡。由於乙醯膽鹼和GABA的減少,造成多巴胺相對增加,引起亨廷頓病患者多動。
亨廷頓病患者痴呆的病理生理學有爭論。有些學者將精神活動減少歸因於大腦皮質改變。但亨廷頓病皮質的神經病理改變不恆定,且患者缺乏具有皮質病變特徵的語言和運用障礙。CT顯示尾狀核萎縮與認知測試結果有關,SPECT也發現尾核區腦血流下降與認知測試結果一致,都提示痴呆主要是皮質下病變的結果。亨廷頓病患者在計畫和組織、變換、詞的流暢性以及記憶搜尋等額葉功能上,有明顯障礙。因尾狀核接受額葉投射,成為額葉系統中一部分。當尾狀核病變時,表現出額葉功能障礙。前額葉皮質與尾狀核的聯繫中斷,是亨廷頓病患者認知障礙的基礎。
診斷
 亨廷頓病性痴呆
亨廷頓病性痴呆本病主要診斷依據是:(1)家族史;(2)中年起病;(3)舞蹈樣症狀進行性加重;(4)進行性痴呆。但也可有散發病例。有些首先出現智慧型低下而無舞蹈樣動作,早期診斷較困難,但如有亨廷頓病家族史亦可確診。反之。只有舞蹈症狀而無痴呆者,早期診斷亦有困難。這類病例往往會被誤診為神經性抽搐,但神經性抽搐是刻板的,而亨廷頓病的舞蹈樣動作則否。實際上在亨廷頓病早期,痴呆症狀雖不明顯,正規的神經心理測驗可發現智商偏低。如有亨廷頓病家族史也可確診。
關於舞蹈病診斷問題,有人在有症狀的亨廷頓病患者中發現皮膚成纖維細胞培育中細胞密度和代謝的需求紊亂,以及淋巴細胞和細胞膜的特性異常,其結果可能對症狀前的人有診斷意義。在正電子放射斷層照相上尾狀核葡萄糖代謝減低,也可能在受累者其他特徵出現之前先發現。有人根據L-Dopa能改善運動遲緩與強直症狀,而過量又會誘發舞蹈樣運動這種作用,以此作為該病的預測試驗,即給有危險的對象用L-Dopa2.5g/日,持續10周,如能引起不自主的舞蹈運動,則可能是該病致病基因攜帶者。這些技術可用作無症狀。但有得病危險者的診斷試驗。但現在尚沒有方法能確切地將有危險但未受累者與已帶有本病基因的患者區分開來。
由於亨廷頓病具有遲發的常染色體顯性遺傳特點,常在中年發病之前就有可能已將致病基因傳給了後代。因此對亨廷頓病的早期診斷具有重要意義。已發現亨廷頓病的基因及其在染色體上的定位,並已證明ITl5基因的不穩定突變是導致中國人亨廷頓病的遺傳學基礎,為本病的早期診斷、遺傳諮詢提供了行之有效的科學方法。
鑑別診斷
亨廷頓病需與良性家族性舞蹈病、小舞蹈病、棘紅細胞增多舞蹈病、肝豆狀核變性、遲發性運動障礙和成人肌張力障礙脂質沉積症相鑑別。
良性家族性舞蹈症(benignfamilialchorea)的典型臨床症狀起始於正常運動發育期後的要兒和幼年。起病後舞蹈為非進行性。但終身存在。此病並無痴呆。已報導此病為遺傳性和家族性,以常染色體顯性和常染色體隱性兩種形式遺傳。
小舞蹈病(sydenhamchorea)的突出臨床特徵是自發的運動不協調和力弱。小舞蹈病主要發病於5-15歲,女性患者為男性的兩倍。首次發病後持續時間不超過六個月,但三分之一患者有復發。動作為暴發、唐突、跳動樣和抽搐樣,而不是典型亨廷頓病者的舞蹈樣運動,抽搐缺乏刻板式。與亨廷頓病比較。在閃電樣抽搐中,累及肌肉較少,且遠端肌肉受累較突出。常見情緒不安和暴躁,痴呆則罕見。可有風濕熱及其它表現,如心肌炎和關節炎,血沉快或抗鏈球菌溶血素滴定度可增高。在有小舞蹈病發作史的病人中,有許多可發生孕婦舞蹈病和口服避孕藥引起的舞蹈病。
與亨廷頓病具有許多共同特點的一種病是家族性基底神經節變性的棘紅細胞增多症——舞蹈棘紅細胞增多症(acanthocytosis—choreoacanthocy—tosls)。此症發病於15~35歲,以舞蹈症和突出的口面運動障礙開始發病。周圍神經病常見。運動障礙導致進行性病殘,於50~70歲死亡。此病散發,或以常染色體隱性遺傳。嚴重的行為障礙和情緒改變與不專心和注意力下降,情感淡漠和概括能力受損一起發生。CT掃描示尾狀核萎縮。血塗片發現5%~15%紅細胞為棘紅細胞。血清肌酸磷酸激酶和乳酸脫氫酶含量可增高。與棘紅細胞增多症的Bassen-Kornzweig綜合徵相反,血清β-脂蛋白不減少。肌電圖和肌肉活體檢查符合神經元性萎縮。屍體解剖檢查證明尾狀核和殼核萎縮,並有小細胞消失。大神經元保存與亨廷頓病的變化相似。但谷氨酸脫氫酶活性並不特別降低。臨床上舞蹈棘紅細胞增多症與亨廷頓病的區別是:遺傳的隱性形式。無明顯痴呆。有神經元性肌萎縮及棘紅細胞增多。
肝豆狀核變性(hepatolenticulardegeneration)可有不自主多動及精神症狀,但有肝功能損害。且大部分病人有角膜K-F環,血清銅藍蛋白明顯減低、血清總銅量低而尿銅增高,用青零胺等治療效果較好可與亨廷頓病鑑別。
遲發性運動障礙(tardivedyskinesia)是指慢性套用神經阻滯劑的人發生運動障礙。最顯著的動作障礙累及口和舌,但手、腿、軀幹和呼吸肌也可發生舞蹈手足徐動症。推測這種病是繼發於基底神經節中多巴胺受體超敏感,而這種超敏感是由於慢性多巴胺受體阻滯而產生的。由於難評價強安定劑和抗膽鹼能藥以及其他各種精神活性藥持續治療的效果,以及某些精神病人晚期發生的智慧型衰退,使識別這種遲發性痴呆複雜化。然而中樞神經系統機制的破壞對遲發性運動障礙起作用,這種機制破壞,也可產生某些認知功能損害。此領域需要更廣泛的研究。以肯定或反證這些初步觀察。診斷依賴於相應神經阻滯劑的藥物史,實驗室檢查無助於遲發性運動障礙的診斷。
成人肌張力障礙脂沉積症(adultdystoInicLipidosis)是很罕見的神經內臟儲存障礙,並有核上性眼肌麻痹,輕度痴呆、舞蹈手足徐動症、肌張力障礙和共濟失調。骨髓中可發現泡沫細胞和海藍組織細胞。
治療
 亨廷頓病性痴呆
亨廷頓病性痴呆對亨廷頓舞蹈病的治療,運動障礙本身如不嚴重一般不需藥物治療反之,如運動幅度大引起摔倒時,可佐以多巴胺耗竭劑如利舍平,但應監測血壓和利舍平引起的抑鬱由於焦慮應激,不自主運動加重時,可用抗焦慮藥苯二氮卓類,如病人肌強勁明顯可用DA激動劑以改善肌強勁。氟哌啶醇、三氟拉嗪氟、奮乃靜等對舞蹈樣症狀精神病樣症有效,但注意上述藥物可引起抑鬱或加重原有的抑鬱症狀。
精神病性症狀可選用對錐體外系副作用較輕的抗精神病藥,劑量宜小通過DA阻斷作用對控制舞蹈樣動作可能有效抑鬱可選用新一代抗抑鬱藥如帕羅西汀、舍曲林、氟西汀對焦慮抑鬱都有效而三環類抗抑鬱藥的抗膽鹼能副作用可能加劇舞蹈症如出現妄想精神病性症狀,可用抗精神病藥,但劑量宜小。甲硫噠嗪、氯氮平因錐體外系反應輕,可能更適宜。
此外,鑒於精神和軀體方面的障礙要對病人做好諮詢解釋,並在以後的發展階段做好護理工作
