
簡介
認知是機體認識和獲取知識的智慧型加工過程,涉及學習、記憶、語言、思維、精神、情感等一系列隨意、心理和社會行為。認知障礙(cognitivedisorder)指與上述學習記憶以及思維判斷有關的大腦高級智慧型加工過程出現異常,從而引起嚴重學習、記憶障礙(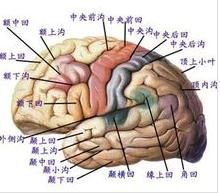 認知障礙
認知障礙主要分類
主要包括(1)感知障礙,如感覺過敏、感覺遲鈍、內感不適、感覺變質、感覺剝奪、病理性錯覺、幻覺、感知綜合障礙;
(2)記憶障礙,如記憶過強、記憶缺損、記憶錯誤;
(3)思維障礙,如抽象概括過程障礙、聯想過程障礙、思維邏輯障礙、妄想等。
上述各種認知障礙的原因是多種多樣的,除器質性疾病原因外,大多精神疾患所致。如神經衰弱、癔症、疑症、更年期綜合症、抑鬱症、強迫症、老年性痴呆、精神分裂症、反應性精神病、偏執型精神病、躁狂症、躁鬱症等等。
具體分類
感知覺障礙
(一)感覺障礙感覺障礙(disordersofsensation)主要包括感覺過敏,感覺減退和內感性不適。
 失語症
失語症(二)知覺障礙
知覺障礙(distuebanceofperception)主要包括錯覺和幻覺。
1、錯覺(illusion)指對客觀事實的歪曲的知覺、生理和病理情況下都可能產生錯覺。
2、幻覺(hallucination)
幻覺是虛幻的知覺指沒有外界相應的客觀刺激作用於感覺器官時所出現的知覺體驗。幻覺是臨床最常見而且重要的精神病性症狀,常與妄想合併存在。
(1)根據所涉及的感覺器官,幻覺可以分為幻聽、幻視、幻嗅、幻味,內臟性幻覺。
(2)按幻覺體驗的來源分為真性幻覺和假性幻覺。
(3)按幻覺產生的特殊條件可分為功能性幻覺、思維鳴響、心因性幻覺和入睡前幻覺。
(三)感知綜合障礙
感知綜合障礙(psychosensorydisturbance)是指患者對客觀事物的本質屬性或整體能正確感知,但對某些個體如大小,形狀。顏色、距離、空間位置等產生錯誤的感知,多見於癲癇。
思維障礙
(一)思維形式障礙(disorderofthethinkingform)包括思維聯想障礙和思維邏輯障礙兩大部分。(二)思維內容障礙
1、妄想(delusion)是一種病理的歪曲信念,是病態的推理和判斷,是精神病患者最常見的症狀之一。其特徵有:
 研究
研究①信念的內容與事實不符,沒有客觀現實基礎,但患者堅信不疑;
②妄想內容均涉及患者本人,與個人利害有關;
③妄想具有個人獨特性;
④妄想內容因文化背景和個人經歷而有所差異,但具有濃厚的時代色彩。
2、強迫障礙(obsessiveidea)又稱強迫思維,是指某一觀念或概念,反覆的出現患者的腦海中,自己知道這種想法是不必要的,甚至是荒謬的,並力圖加以擺脫。但事實上常常視為被患者的意願,想擺脫,有擺脫不了,患者為此而苦惱。
3、超價觀念(over-valuedidea)是在意識中占主導地位的錯誤觀念,其發生常常有一定的事實基礎,但患者的這種觀念是片面的,與實際情況有出入,而且帶有強烈的感情色彩,明顯的影響到患者的行為。
注意障礙
注意(attention)是指個體的精神活動集中地指向於一定對象的過程。此時,人們對所注意的食物的感知最為清晰,而對周圍其他事物的感知相對不清晰。記憶障礙
智慧型障礙
1、精神發育遲緩(mentairetardation)也稱智力低下,是指先天或圍生期或在生長發育成熟以前(18歲以前),由於多種致病因素,遺傳,感染,中毒,頭部外傷,內分泌異常或缺氧等,是大腦發育不良或發育受阻,一直智慧型發育停留在某一階段。不能隨著年齡增長而增長,其智慧型明顯低於正常同齡人。2、痴呆(dementia)是一種綜合症,是意識清楚情況下後天獲得的記憶,智慧型的明顯受損。
自知障礙
自知力(insight)又稱領悟力或內省力,是指患者對自己精神疾病的認識和判斷能力。認知的腦結構基礎
認知的結構基礎是大腦皮層。大腦皮層由主區(primarycortex)和輔助區(associatedcortex)組成,對事物的觀察、分析與判斷以及對軀體運動的協調均由主區控制,但主區完成這些功能依賴輔助區對行為和認知障礙
智慧型進行高層次整合。
Brodmann根據形態特徵將大腦皮層分為52個功能區,並提出不同的皮層形態分區分別執行不同的功能。①額葉皮層區負責自主運動,書寫、記憶、創造性思維、判斷、遠見、社會責任感等複雜的智力活動,該區損傷將導致中側性偏癱(4區)、失寫症(6區)及額葉性痴呆(9區和12區)等;腦左半球額葉皮層Broca’s語言區(44區和45區)損傷導致運動性失語症;②頂葉皮層的主要功能是對感覺信息的高級加工和整合。頂葉皮層l區至3區的損傷導致對側感覺障礙;39區的損傷導致感覺性失讀症(此時患者無構語障礙,但不能理解書寫的文字);40區的損傷引起觸覺缺失等;③顳葉接受聽覺刺激,其4l區和42區感受聲音,而聽覺輔助皮層22區幫助對聲音的理解,22區損傷將導致感覺性(Wernicke’s)失語症(與Broca’s失語症不同,Wernicke’s失語症者不能正確使用語言和語法,常常言不達意);顳葉的海馬和藍斑結構參與記憶加工。損傷時分別引起空間或情感記憶障礙;④枕葉含有原始視覺皮層,17區感知和接受視覺刺激,該區損傷引起視野缺陷;視覺聯絡皮層18區和19區包繞視皮層,詮釋視覺信息和內容。該區損傷將導致個體不能識別物體,不理解物體的用途或生命的形式(如不能區別貓和狗)。
病因及發病機制
認知是大腦皮層複雜高級功能的反映,任何直接或間接導致大腦皮層結構和功能慢性損傷的因素均可通過不同機制引起認知障礙,現將其歸納如下:慢性腦損傷
1.腦組織調節分子異常(1)神經遞質及其受體異常:大多數神經元之間的信息傳遞是通過神經遞質(neurotransmitter)及其相應的受體完成的。這些神經遞質或受體異常改變均可導致不同類型和不同程度的認知異常。
1)多巴胺(dopamine):多巴胺是以酪氨酸為底物,在酪氨酸羥化酶(tyrosinehydroxylase)和多巴脫羧酶(dopaminedecarboxylase)的作用下合成的。研究發現:腦中多巴胺含量顯著降低時可導致動物智慧型減退、行為情感異常、言語錯亂等高級神經活動障礙。例如,在帕金森病(Parkinsondisease,PD)患者黑質多巴胺能神經元減少,酪氨酸羥化酶和多巴脫羧酶活性及紋狀體多巴胺遞質含量明顯卞降。此外,在動物實驗中發現多巴胺過多也可導致動物認知功能的異常改變。多巴胺受體有D1和D2受體兩大家族,精神分裂症患者與大腦額葉皮層的D1受體功能低下和皮層下結構D2受體功能亢進雙重因素有關,因此有人提出用D1激動和D2阻斷治療精神分裂症的新概念。
2)去甲腎上腺素(nonepinephrine):去甲腎上腺素是最早被發現的單胺類神經遞質,是多巴胺經β羥化酶作用生成的產物。在腦內,去甲腎上腺素通過α1、α2和β受體發揮調節作用。在突觸前,α2受體通過Gi蛋白介導,減少cAMP的生成和cAMP依賴性蛋白激酶的活性,減少蛋白激酶對N-型Ca2+通道的磷酸化,以至Ca2+通道關閉,Ca2+內流減少,從而對去甲腎上腺素的釋放起抑制作甩(負反饋調節);α2受體激動還可抑制在警醒狀態下的藍斑神經元的放電增加;在突觸後,α1受體激動可引起K+通道開放,K+外流增加,神經元傾向超極化而產生抑制效應。而α1受體激活則使K+通道功能降低,K+外流減少,神經元去極化產生興奮效應。一般認為,腦中α2受體激動與維持正常的認知功能有關,而α1受體持續、過度激活可致認知異常。在正常警醒狀態時,腦細胞含適量去甲腎上腺素,α2受體功能占優勢,維持正常的認知功能。在應激狀態下產生大量去甲腎腎上腺素,α1受體功能占優勢;這可能是個體長期處於應激狀態更易出現認知障礙的機制之一。
3)乙醯膽鹼(aeetylcholine):乙醯膽鹼由乙醯輔酶A和膽鹼在膽鹼乙醯轉移酶的作用下生成。神經細胞合成並釋放的乙醯膽鹼通過M-受體(M-AchR,毒蕈鹼受體)和N-受體(N-AchR,菸鹼受體)發揮調節作用,M-AchR是G-蛋白耦聯受體,N-AchR是配體門控離子通道受體。腦內的膽鹼能神經元被分為兩類,即局部環路神經元和投射神經元,自Meynert基底核發出的膽鹼能纖維投射至皮層的額葉、頂葉、顳葉和視皮層,此通路與學習記憶功能密切相關。阿爾茨海默病(Alzheimer'sdisease,AD)患者在早期便有Meynert基底區膽鹼能神經元減少,導致皮層膽鹼乙醯轉移酶活性和乙醯膽鹼含量顯著降低,是AD患者記憶障礙的重要機制之一;精神分裂症者認知障礙的程度與皮層膽鹼乙醯轉移酶活性呈負相關;給AD和精神分裂症患者使用膽鹼酯酶抑制劑或M受體激動劑可改善其記憶缺損。
4)谷氨酸(glutamate):在腦內,胺基酸類遞質含量最高,其中,谷氨酸在人大腦皮層中的含量約為9-11μmol/g,比乙醯膽鹼或單胺類遞質的含量高103數量級,比神經肽的含量高106數量級。谷氨酸是不能透過血腦屏障的非必需胺基酸,腦內的谷氨酸可分別由谷氨醯胺在谷氨醯胺酶的作用下水解或α-酮戊二酸在其轉氨酶的作用下生成。谷氨酸藉N-甲基-D-門冬氨酸(N-methyl-D-aspartate,NMDA)和非NMDA受體起作用。NMDA受體是配體門控的離子通道型受體;非NMDA受體主要指海人藻酸(kainate,KA)和α-氨基-3-羥基-5-甲基-4-異惡唑-丙酸(α-mino-3-hydroxy-5-methy-4-isoxa-zolep-propionate,AMPA)是Na+-K+通透性離子通道型受體。紋狀體的谷氨酸神經纖維抑制丘腦向大腦皮層發出感覺衝動,當谷氨酸能神經低下時,這種衝動發出增多,大腦皮質單胺活性增強,引起相應的認知功能異常。由於谷氨酸是哺乳動物腦內最重要的興奮性神經遞質,故當谷氨酸含量異常增高時,可引起“興奮性毒性”損傷。
(5)神經肽異常:神經肽(neuropeptide)是生物體內的一類生物活性多肽,主要分布於神經組織。在腦內,神經肽與神經遞質(neurotransmitter)常常共存於同一神經細胞,但神經肽與經典神經遞質有諸多不同:神經肽比神經遞質分子量大,在腦組織中含量低;神經肽由無活性的前體蛋白加工而成,而神經遞質可在胞體或神經末梢直接合成;神經肽釋放後主要經酶解而失活,神經遞質則主要通過神經末梢重吸收反覆利用;神經肽的調節緩慢而持久,神經遞質的調節快速而精確等。神經肽的異常與認知障礙密切相關。有人報導PD患者腦蒼白球和黑質中P物質水平下降30%-40%,在黑質中膽囊收縮素(cholecystokinin,CCK)下降30%,在丘腦下部和海馬區神經降壓肽(neurotensin,NT)含量也下降。血管加壓素(vasopressin,VP),血管活性腸肽(vasoac-tireintestinalpeptide,VIP)及其受體含量減少與記憶力減退相關,給腦外傷、慢性乙醇中毒及AD病人用VP可改善其記憶力減退。促甲狀腺素釋放激素(thyrotropinreleasinghormone,TRH)是第一個從丘腦下部分離出來的三肽激素,TRH可引起行為改變,如興奮、精神欣快及情緒暴躁等。TRH既可以作為一種神經激素通過受體調節其他遞質起作用,又可以作為一種神經遞質直接起作用。腺垂體分泌的促腎上腺激素釋放激素(adrenocorticotropichormone,ACTH)是一39肽激素,其水平改變影響動物的學習記憶、動機行為等。ACTH影響動物學習和行為的關鍵分子區域是其分子中第4——10位胺基酸殘基,該片斷能提高大鼠的注意力和記憶力,同時減輕動物的焦慮行為。多發性硬化(multiplesclerosis,MS)患者丘腦下部-垂體一腎上腺皮質(hypothalamus-pynear-adrenocorticode,HPA)軸功能紊亂與其反應遲鈍、智慧型低下、重複語言等認知功能障礙顯著相關。根據絕經期女性AD的發病率高於男性,且經絕後接受雌激素替代療法者的患病率降低,有人提出性激素代謝紊亂也可能參與認知障礙的發病過程。
(6)神經營養因子缺乏:神經元和膠質細胞可合成、分泌大量的神經營養因子,如神經生長因子(neurogrowthfactor,NGF)、睫狀神經營養因子(ciliaryneurotrophicfactor,CNTF)、腦源性神經營養因子(brain-derivedneurotrophicfactor,BDNF)和膠質源性神經營養因子(glia-derivedneu-rotrophicfactor,GDNF)等。這些神經營養因子對神經元的存活和神經元突起的生長具有重要作用。已發現在多種神經退行性疾病中均有神經營養因子含量的改變,例如,在PD患者黑質NGF、BDNF和GDNF的含量明顯降低,離體和在體實驗均證明BDNF、GDNF和CNTF對吡啶類衍生物1-甲基4一苯基l,2,3,6一四氫吡啶(MPTP)造成的多巴胺能神經元損傷具有很強的保護作用。
2.腦組織蛋白質異常聚集腦組織中蛋白質異常聚集可見於一大類腦神經細胞退行性變性疾病中,如AD、PD、亨廷頓病(Huntingtondisease,HD)、海綿狀腦病(CreutzfeldtJokobdisease,CJD)等。蛋白質的異常聚積與基因變異、蛋白質合成後的異常修飾、腦組織慢病毒感染、腦老化和環境毒素中毒等多種因素有關。
(1)基因異常:已發現多種基因異常參與神經細胞的退行性變性。例如,在PD患者有ot-synuclein,parkin和park3基因突變,a-synuclein基因第209位的核苷酸發生了G-A錯義突變,使其蛋白質第53位的丙氨酸(Ala)變成了蘇氨酸(Thr),變異的蛋白質是PD患者神經細胞胞漿中特徵性嗜酸性包涵體,即路易(Lewy)小體的重要成分;已發現有30多種不同parkin基因缺失和點突變與早發性PD有關,改變的parkin蛋白可導致依賴泛素的蛋白降解過程異常,促使parkin蛋白聚集。在AD患者,已發現5個相關基因突變,所編碼的蛋白質依次為澱粉樣前體蛋白(amy-loidprecursorprotein,APP)、早老蛋白-1(presenilin-1,PS-1)、PS-2、載脂蛋白E(apolipoproteinE,apoE)和α2-巨球蛋白(α2-macro谷氨酸bumin)。其中,APP、PS基因突變和ApoE基因多態性可導致APP異常降解,產生大量B澱粉樣多肽(AB),過量產生的Ap不斷在神經細胞間聚集形成老年斑,同時可導致過氧化損傷(損傷生物膜、破壞細胞內鈣離子穩態、抑制星形膠質細胞、使一些關鍵酶失活)、炎症反應和神經細胞死亡。
(2)蛋白質合成後的異常修飾:正常時,蛋白質合成後的不同加工修飾賦予蛋白質不同的結構和功能,是蛋白質結構和功能多樣性的基礎。蛋白質的異常修飾導致其結構異常、功能降低或喪失。在AD患者,發現細胞骨架蛋白tau被異常磷酸化(phosphorylation)、異常糖基化(glycosylmion,酶促反應)、異常糖化(glycmion,非酶促反應)和異常泛素化(ubiquitilation)修飾,異常修飾的tau蛋白沉積在神經細胞中形成神經原纖維纏結。關於tau蛋白異常糖基化、異常糖化和異常泛素化的機制尚不清楚,目前認為AD患者tau蛋白被異常磷酸化可能與蛋白磷酸酯酶(proteinphosphatase)和蛋白激酶(proteinkinase)調節失衡有關。蛋白磷酸酯酶催化蛋白質去磷酸化,AD患者腦中蛋白磷酸酯酶的活性明顯降低,使tau蛋白去磷酸化減弱,導致AD患者腦中tau蛋白異常過度磷酸化。蛋白激酶催化蛋白質磷酸化,在AD患者,大腦顳葉皮層多種蛋白激酶的表達量或活性比對照者顯著增強。上述磷酸化系統失衡導致tau蛋白異常過度磷酸化,異常修飾的tau在神經細胞內聚集是AD患者神經細胞退化的重要機制。
(3)腦組織慢病毒感染:最常見的由慢病毒感染引起的人類中樞性疾病為CJD,是由一種具傳染性的朊蛋白(prionprotein,PrP)所致。這種PrP類似於病毒可傳播疾病,但與已知病毒不同是,它沒有任何可檢測到的核酸序列。人類PrP蛋白有兩種異構體,分別是存在於正常細胞的PrP(PrPc)和引起朊蛋白病的PrPsc(PrPscrapie)。兩種異構體的序列並無差別,但蛋白質的空間構型不同。PrPc是一種細胞內膜結合蛋白,PrPsc不僅存在於細胞內膜,還存在於朊蛋白病患者神經細胞外的澱粉樣蛋白纖絲和斑塊中;prpsc可促進PrPc轉化為PrPsc。在人體內,PrPsc的增殖是通過一分子PrPc與一分子PrPsc結合形成雜二聚體,此二聚體再轉化成兩分子PrPsc,PrPsc便依此呈指數增殖。有朊蛋白基因突變時,細胞中的PrPc。更易從α-螺鏇轉變成β-片層,此時更容易與PrPsc結合,導致PrPsc增殖和聚集。
3.慢性腦缺血性損傷
神經元能量儲備極少,對缺血、缺氧非常敏感,完全缺血5分鐘即可導致神經元死亡。腦缺血造成大腦皮層損傷是引起不同類型認知障礙的常見原因。統計資料表明:腦卒中患者在發病後出現痴呆的危險性較同齡對照組明顯增高;有腦卒中史的老年群體的認知水平亦低於無卒中史的同齡老人。腦細胞缺血引起認知異常的機制可能與下述因素有關。
(1)能量耗竭和酸中毒:在缺血、缺氧狀態下,細胞的能量代謝轉為無氧酵解。無氧酵解生成ATP的效率低,使細胞出現能量耗竭。無氧酵解引起腦組織缺血性乳酸酸中毒,細胞Na+-K+泵功能損傷,K+大量外溢,同時Na+、Cl-及Ca2+大量流人細胞內引起細胞損傷;缺血區乳酸堆積還可引起神經膠質和內皮細胞的水腫和壞死,加重缺血性損害。
(2)細胞內Ca2+超載:腦缺血時,神經細胞膜去極化,引起大量神經遞質釋放,興奮性遞質(如谷氨酸)的釋放激活NMDA受體,使鈣通道開放,Ca2+內流增加;如激活非NMDA受體,使Ca2+從內質網釋放至細胞漿內;膜去極化本身也啟動了電壓依賴性鈣通道,加重Ca2+內流。神經細胞Ca2+超載可通過下述機制導致細胞死亡:①Ca2+超載時,大量Ca2+沉積於線粒體,干擾氧化磷酸化,使能量產生障礙;②激活細胞內Ca2+依賴性酶類,其中Ca2+依賴的中性蛋白水解酶過度激活可使神經細胞骨架破壞;③激活磷脂酶A和磷脂酶C,使膜磷脂降解;產生大量游離脂肪酸,特別是花生四烯酸,後者在代謝過程中產生血栓素、白三烯,一方面通過生成大量自由基加重細胞損害;另一方面可激活血小板,促進微血栓形成,在缺血區增加梗死範圍,加重腦損害;④腦缺血時,腦血管平滑肌,內皮細胞均有明顯Ca2+超載,前者可致血管收縮、痙攣,血管阻力增加,延遲再灌流,使缺血半暗帶內側支循環不能形成,從而腦梗死灶擴大;後者可致內皮細胞收縮,內皮間隙擴大,血腦屏障通透性增高,產生血管源性腦水腫。
(3)自由基損傷:在急性腦缺血時,自由基產生和清除平衡狀態受到破壞而引起腦損傷。其機制為:①缺血腦細胞能量衰竭,谷氨酸、天門冬氨酸(Asp)增多,此時電壓依賴性鈣通道和NM.DA受體操縱的鈣通道開放,鈣離子大量內流,使黃嘌呤脫氫酶轉化為黃嘌呤氧化酶,後者催化次黃嘌呤氧化為黃嘌呤並同時產生氧自由基;鈣離子大量內流還可激活磷脂酶A,造成血管內皮細胞和腦細胞的膜磷脂降解,花生四烯酸產生增加,後者代謝產生自由基;②缺血區腦細胞線粒體內鈣離子增多,三羧酸循環發生障礙,不能為電子傳遞鏈的細胞色素氧化酶提供足夠的電子將O2還原成H2O,從而生成氧自由基,並漏出線粒體;③急性腦缺血時,NO增多,NO能與氧自由基相互作用形成過氧亞硝基陰離子,後者又分解成羥自由基(OH-)和二氧化氮自由基(NO2-);④梗死灶內游離血紅蛋白和鐵離子與存在於細胞內的H202發生反應,產生OH-和氧自由基。兒茶酚胺等物質亦可發生氧化反應生成氧自由基。⑤缺血灶由於趨化因子增加,在血管內皮表面吸附大量中性粒細胞和血小板,前者通過細胞色素系統和黃嘌呤氧化酶系統產生O氧自由基和H202,後者通過血小板活化因子引起細胞內Ca2+濃度升高,促進自由基生成。
(4)興奮性毒性:中樞神經系統中大部分神經遞質是胺基酸類,包括谷氨酸、天冬氨酸、γ-氨基丁酸(GABA)和甘氨酸。其中,谷氨酸和天冬氨酸對神經元有極強的興奮作用,故稱為興奮性胺基酸(excitatoryaminoacid,EAA),GABA和甘氨酸對神經元行使抑制作用,故稱為抑制性胺基酸(inhibitoryaminoacid,IAA)。“興奮性毒性(excitatorytoxicity)”指腦缺血缺氧造成的能量代謝障礙直接抑制細胞質膜上Na+-K+-ATP酶活性,使胞外K+濃度顯著增高,神經元去極化,EAA在突觸間隙大量釋放,因而過度激活EAA受體,使突觸後神經元過度興奮並最終死亡的病理過程。EAA通過下述兩種機制引起“興奮性毒性”:一是AMPA受體和KA受體過度興奮引起神經細胞急性滲透性腫脹,可在數小時內發生,以Na+內流,以及Cl-和H2O被動內流為特徵;另一種是NMDA受體過度興奮所介導的神經細胞遲發性損傷,可在數小時至數日發生,以持續的Ca2+內流為特徵。
認知障礙的病因及發病機制
(5)炎症細胞因子損害:在腦缺血損害發生後,產生多種多效性細胞因子。在致炎細胞因子占主導地位時,加重腦缺血損害,在抗炎因子占主導時,對腦缺血產生保護作用。如白細胞介素-1β(IL-1β)和腫瘤壞死因子-α(TNF-α)加重腦缺血損害,轉化生長因子β1(TGFβ1)對腦缺血有保護作用。此外,在缺血損傷的神經元釋放的細胞因子激發下,缺血區吞噬細胞明顯增加,吞噬細胞既能釋放細胞因子刺激修復過程,又可釋放神經毒素殺傷存活神經元。
4.環境、代謝毒素對腦的損害
對絕大多數50歲以後發病的典型散發性神經退行性疾病而言,環境和代謝毒素對腦的損害起主要作用,這些風險因素包括毒品、藥物、酒精或重金屬中毒等。各種慢性代謝性或中毒性腦病時,如心肺衰竭、慢性肝性腦病、慢性尿毒症性腦病、貧血、慢性電解質紊亂、維生素B:缺乏、葉酸缺乏等,其主要表現為認知異常。
5.腦外傷
腦外傷對學習記憶和智力有不同程度的影響。輕度外傷者可不出現症狀;中度外傷者可失去知覺;重度者可導致學習記憶嚴重障礙,乃至智力喪失。例如,一些“被打得昏頭轉向”的拳擊手,腦反覆損傷可出現構語障礙(口吃),心不在焉,好爭辯,注意力渙散,近期記憶減退,步態僵硬、痙攣等。
6.腦老化
認知功能一般隨年齡增高(約60歲以後)而下降。研究發現,PD患者黑質多巴胺能神經元、酪氨酸羥化酶和多巴脫羧酶活力、紋狀體多巴胺遞質自30歲以後隨年齡增長而逐年減少或降低。老年人腦巾血液供應減少,台成和分解代謝以及對毒素的清除能力均降低,這些都是造成老化腦神經細胞死亡,認知功能降低的主要因素。
慢性全身性疾病
心血管系統病變,如高血壓、糖尿病、慢性阻塞性肺疾病等,可通過減少腦血液供應等機制,繼發性降低大腦功能而引起認知障礙。處於亞臨床階段的心、腦血管疾病的高危人群,其認知測驗的得分明顯低於無任何亞臨床特徵的同齡老人,說明這些病變可能已經造成腑部的缺血、缺氧及腦功能損傷。此外,整體功能水平降低,如老年人聽力下降使其與外界環境的接觸以及對外界刺激的加工減少,也可降低老年人對外界環境的感知和認同;軀體功能,特別是操作性活動減少也可導致認知功能減退。有人發現,冠脈搭橋手術後的患者常出現短期記憶喪失和注意力下降,還有人認為,任何一種大的外科手術都可能導致大腦皮層功能的上述改變。精神、心理異常
輕鬆、愉快、多彩的生活環境可促進實驗動物大腦皮層的增長,使腦重量增加。相反,不良的心理、社會因素,如負性生活事件、處境困難、驚恐、抑鬱等均可成為認知障礙的誘囡。近年來,利用電子計算機x線斷層掃描(CT)與磁共振(MRI)對精神活動失調患者的腦成像研究發現,社會心理功能減退患者的有關腦區的皮層萎縮。用正電子發射掃描(PET)和單光子發射計算機斷層掃描(SPECT)結契約位索示蹤對腦局部腦血流(rCBF)和18氟一脫氧葡萄糖(FDG)或11碳-脫氧葡萄糖(CDG)代謝的研究證實,精神失常患者的有關腦區局部血流低灌注,葡萄糖利用率降低。用電子顯微鏡觀察並經圖像分析發現,精神分裂症患者的有關腦區神經細胞數目減少,細胞體積變小。人文因素的影響
在諸多的人文因素中,受教育程度是報告最多、結果最恆定的影響認知的因素,認知測驗的得分與受教育年限呈負相關。社會地位低下,經濟生活狀況較差與認知功能減退和痴呆的發生有一定關係。但在多因素分析中控制了年齡、性別、卒中史等較重要的因素後,社會經濟凼素的影響一般不再顯著。此外,女性認知功能損害的發生率高於男性,對各年齡組進行多因素分析的結果表明,這種差異與女性的受教育程度較低和慢性病患病率較高有關。圖18-2簡要概括認知障礙的病因和發病機制。6主要表現形式
形式概況
人腦所涉及的認知功能範疇極其廣泛,包括學習、記憶,語言、運動,思維、創造,精神、情感,等等,因此,認知障礙的表現形式也多種多樣,這些表現可單獨存在,但多相伴出現。學習、記憶障礙
學習、記憶是一種複雜的動態過程,對學習、記憶基本機制的了解得益於對一種低等無脊椎動物海兔(aplysia)的簡單的神經系統的研究。記憶是處理、貯存和回憶訊息的能力,與學習和知覺相關。記憶過程包括感覺輸入→感覺記憶→短時記憶→長時記憶→貯存訊息的回憶等過程。短時記憶涉及特定蛋白質的磷酸化和去磷酸化平衡,而長時記憶除特定蛋白質的磷酸化改變外,還涉及新蛋白質的合成。在大腦皮層不同部位受損傷時,可引起不同類型的記憶障礙,如顳葉海馬區受損主要引起空間記憶障礙,藍斑、杏仁核區受損主要引起情感記憶障礙等。失語
失語是由於腦損害所致的語言交流能力障礙。患者在意識清晰、無精神障礙及嚴重智慧型障礙的前提下,失語症
無視覺及聽覺缺損,亦無口、咽、喉等發音器官肌肉癱瘓及共濟運動障礙,卻聽不懂別人及自己的講話,說不出要表達的意思,不理解亦寫不出病前會讀、會寫的字句等。傳統觀念認為,失語只能是由大腦皮層語言區損害引起。CT問世後證實,位於優勢側皮層下結構(如丘腦及基底節)病變也可引起失語。失認
失認是指腦損害時患者並無視覺、聽覺、觸覺、智慧型及意識障礙的情況下,不能通過某一種感覺辨認以往熟悉的物體,但能通過其他感覺通道進行認識。例如,患者看到手錶而不知為何物,通過觸摸手錶的外形或聽表走動的聲音,便可知其為手錶。失用
要完成一個複雜的隨意運動,不僅需要上、下運動神經元和錐體外系及小腦系統的整合,還須有運動的意念,這是聯絡區皮層的功能。失用是指腦部疾患時患者並無任何運動麻痹、共濟失調、肌張力障礙和感覺障礙,也無意識及智慧型障礙的情況下,不能在全身動作的配合下,正確地使用一部分肢體功能去完成那些本來已經形成習慣的動作,如不能按要求做伸舌、吞咽、洗臉、刷牙、劃火柴和開鎖等簡單動作,但病人在不經意的情況下卻能自發地做這些動作。一般認為,左側緣上回是運用功能的皮層代表區,由該處發出的纖維至同側中央前回,再經胼胝體而到達右側中央前回。因此左側頂葉緣上回病變可產生雙側失用症,從左側緣上回至同側中央前回間的病變可引起右側肢體失用,胼胝體前部或右側皮層下白質受損時引起左側肢體失用。其他精神、神經活動的改變
患者常常表現出語多嘮叨、情緒多變,焦慮、抑鬱、激越(agitation)、欣快等精神、神經活動方面的異常改變。痴呆
痴呆(dementia)是認知障礙的最嚴重的表現形式,是慢性腦功能不全產生的獲得性和持續性智慧型障礙綜合徵。智慧型損害包括不同程度的記憶、語言、視空間功能障礙、人格異常及其他認知(概括、計算、判斷、綜合和解決問題)能力的降低,患者常常伴有行為和情感的異常,這些功能障礙導致病人日常生活、社會交往和工作能力的明顯減退。防治的病理生理基礎
對認知障礙的防治必須根據其病因和發病機制,採用相應的策略。研究
