概述
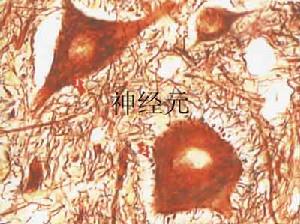 神經元系統
神經元系統神經元蠟樣脂褐質沉積症(neuronalceroidlipofuscinosis,NCL)是一組兒童最常見的遺傳性進行性神經系統變性病。雖然多數患者在兒童期發病,偶爾也出現在成年人其臨床特點:包括進行性痴呆,難治性癲癇。發作和視力喪失在病理上,表現為具有黃色自發螢光,特性的脂色素沉積在神經細胞和其他細胞內,導致以大腦皮質和視網膜為主的神經細胞脫失。超微結構檢查發現脂色素在不同的臨床亞型由顆粒狀、線狀和指紋體狀物質構成。這些沉積物除在中樞神經系統的神經細胞記憶體在外,可以在皮膚活檢和血淋巴細胞的超微結構檢查中發現。
病因病理
 基因
基因是常見的遺傳性進行性神經系統變性病,多數患者在兒童期發病,偶爾也出現在成年人。對NCL的描述可以追溯到1826年,Stengel對此病進行了首次描述並提出了蠟樣質脂褐素沉積病的診斷。1963年Zeman和Alpert在家族性黑蒙性痴呆患者,發現患者腦內沉積物具有黃色自發螢光的特點,從而區別於其他的代謝蓄積性疾病。以後的研究又有不同的命名,同義詞包括:Santavuori-Haltia-Hagberg病Jansky-Bielschowsky病Spielmeyer-Sjögren病,Kufs病及Batten病。Batten病有兩個內涵,一是特指青少年型的NCL,另一個是泛指所有的兒童型NCL,1969年Zeman和Alpert把此病命名為神經元蠟樣脂褐質沉積症,這一病名除美國和英國外已經被廣泛套用。
根據發病年齡病程超微病理,改變和基因異常NCL分4個主要的亞型和4個地區性的變異型:嬰兒型,晚期嬰兒型,青少年型和成年型,除此之外還有一些少見亞型約占所有NCL患者的12%~20%,主要是晚期嬰兒型的4個變異型,包括芬蘭變異型,葡萄牙變異型,土耳其變異型,和進行性癲癇伴智慧型遲緩型嬰兒型,和晚期嬰兒型屬於急性病的範疇,而青少年型和成年型屬於慢性病。
症狀
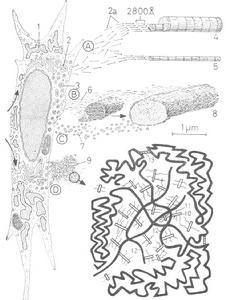 組織細胞
組織細胞1、NCLs主要組織細胞病變特徵為溶酶體內脂質沉積,且沉積物中包含的主要蛋白質為線粒體ATP酶亞基C(除NCL1中SAP A和D)。目前認為NCLs至少由8種不同的基因發生突變引起(CLN1-8)。分子基因學研究已經鑑定並克隆出此疾病群中與6種類型對應的6個基因(CLN1-3,CLN5-6,CLN8)
2、其中CLN8位於常染色體8p,包括3個外顯子,編碼區全長861bp,分為外顯子2和3。
3、 迄今為止,只發現有一個錯義突變(70C->G),導致外顯子2中R24G。
4、 其突變導致進行性癲癇伴智力障礙,其編碼蛋白CLN8P位於內質網和內質網與高爾基體中間連線〔5)為跨膜蛋白,包括286個胺基酸,至少5個跨膜域,具有疏水性,其功能及功能調節機制目前尚不清楚。基於此,我們擬採用酵母雙雜交技術,篩選人胎腦cDNA文庫中與CLN8相互作用的分子。本實驗構建了CLN8基因酵母雙雜交誘餌質粒,並檢測了其對報告基因LacZ的自激活作用。
流行病學
 神經元蠟樣脂褐質沉積症
神經元蠟樣脂褐質沉積症NCL的發病率為每10萬新生兒中出現1.2個患者,不同的國家和地區其發病率不一樣,NCL的不同亞型發病頻率在不同國家和地區也不盡相同,在芬蘭嬰兒型較常見義大利晚期嬰兒型常見,而在德國以青少年型居多,明確診斷的患者也是以青少年型居多不同亞型的發病年齡詳見概述表1。本病具有隱性遺傳的特點,偶爾在成年型NCL出現顯性遺傳,但僅有大約20%的患者有家族史14%的家族可以出現2個患病的兒童,一個家族出現3個和4個患者分別為3.2%和1.17%。
發病機制
 染色體
染色體在NCL的8個分型中目前已經有6個亞型發現基因異常這些基因編碼的蛋白有兩種,一種是溶酶體蛋白酶另一種是膜蛋白。CLNl基因位於常染色體1p32,在223A→G和451C→T發生突變該基因編碼溶酶體酶棕櫚醯蛋白硫脂酶(PPTT1),這種NCL的病理特點是嗜鋨性顆粒沉積,以出現嗜鋨性顆粒為特點的青少年型NCL的致病基因也是編碼PPTTl的基因。CLN2的基因位於常染色體11p15在523→1G→C和636C→T發生突變編碼胃蛋白酶抑制素不敏感的溶酶體肽酶(三肽醯肽酶,TPPl)。CLN3基因位於常染色體16p12該基因發生至少23個突變或1.02-kb的缺失編碼一種功能不明的438胺基酸的跨膜蛋白,命名為Battenin。CLN2的基因位於常染色體13q22可能編碼一種膜蛋白,CLN6的基因位於常染色體15q21-23編碼蛋白不明確CLN8的基因位於常染色體8p23,可能編碼一種286個胺基酸的跨膜蛋白CLN4和CN7的基因目前還不明確。
生化檢查發現在CLNl存在溶酶,體酶,棕櫚醯,蛋白,硫脂酶缺乏,在CLN2存在三肽醯肽酶在CLNl的沉積物中存在鞘脂活性蛋白,CLN2的沉積物含有鞘脂活性蛋白,和線粒體ATP合成酶C亞單位。CLN3CLN4CLN5、CLN6和CLN8沉積物中的主要成分是線粒體ATP合成酶C亞單位。ATP合成酶C亞單位出現線上粒體和溶酶體膜上,它的賴氨酸末端出現甲基化而導致形成儲存型的ATP合成酶C亞單位,肌肉線粒體呼吸鏈的功能在青少年型NCL的沒有明顯的改變,線粒體ATP合成酶C亞單位在細胞內的沉積,可能和該蛋白不能被溶酶體酶正常分解代謝,所致在疾病狀態下作為代謝旁路,泛素溶酶體外蛋白降解系統,線上粒體ATP合成酶C亞單位的分解可能發揮重要的代償作用。
病理改變
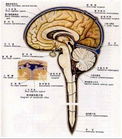 中樞神經系統
中樞神經系統1.中樞神經系統NCL表現為具有黃色自發螢光特性的脂色素,廣泛沉積在神經細胞和其他細胞的胞質內,從而導致神經的氣球樣腫脹,和以大腦皮質及視網膜為主的神經細胞脫失病變,在不同的亞型存在明顯的差異,在嬰兒型和晚期嬰兒型,NCL大腦和小腦皮質的神經細胞脫失很完全,這些改變導致大腦皮質的顯著萎縮和半卵圓中心的有髓神經纖維脫失,脂色素出現在膠質細胞和血管上,皮細胞內晚期嬰兒型NCL的病理改變在不同患者間存在較大的變異,在晚期嬰兒型NCL的Wisniewski變異型出現廣泛的腦萎縮,腫脹的神經細胞主要出現在齒狀核-黑質-紋狀體-丘腦區域,較少出現在大腦皮質。而NCL的色素變異型的中央灰質可見軸突球形成。一例晚期嬰兒型NCL的中央灰質和腦皮質可以同等程度受到明顯的累及,而且腫脹的神經細胞突起也出現在小腦分子層內。
在青少年型NCL神經細胞的脫失不很顯著主要是第Ⅱ層的小神經細胞的喪失,100µm的厚片顯示小神經細胞的喪失和脂色素在神經細胞核周體的出現以皮質Ⅲ和Ⅴ層更明顯,其他神經細胞可以大量存在成年型NCL的神經細胞脫失也不明顯主要表現為皮質和基底核的累及,這些改變常合併有軸突近端的脂色素增加形成軸突的梭形腫脹。北方癲癇型的大腦皮質病理改變也比較輕微小腦沒有明顯的病理改變。
2.視網膜另一組首先受到累及的細胞群,是視網膜的感光細胞可以出現在幾乎所有的兒童型NCL,但成年型NCL和個別青少年型NCL沒有明顯的視網膜病,病變開始於感光細胞層出現進行性的視網膜色素變性,導致整個視網膜萎縮、視網膜血管狹窄和色素從視網膜色素細胞上脫失,視網膜病隨著病情的發展從視網膜周邊向中心發展。
3.軀體非神經細胞軀體細胞也受到脂色素沉積的影響特別是各種腺體細胞。和其他研究相同,研究資料也表明在晚期嬰兒型NCL的肝臟胰腺、腎上腺也存在大量的脂色素沉積,而心肌細胞和結締組織一般較少出現沉積物,皮膚小汗腺分泌部也受到累及。和中樞神經系統不同,軀體非神經細胞累及後沒有臨床表現。
4.超微結構改變在NCL的超微結構檢查可以發現5種不同的嗜鋨性脂褐素顆粒,包括常規脂褐素、顆粒脂褐素、曲線體指紋體和微管聚集。單獨依靠脂褐素的出現不能對NCL進行可靠的分型,但在NCL不同亞型的表現形式是不一樣的,顆粒脂色素出現在嬰兒型NCL,線樣體和指紋體分別出現在晚期嬰兒型和青少年型,極少有顆粒脂色素出現在青少年型患者,成年型NCL為混合型脂褐素顆粒。血淋巴細胞和小汗腺分泌部上皮細胞的超微結構檢查更有助於NCL的診斷和分型,在小汗腺分泌部上皮細胞和淋巴細胞內顆粒脂色素出現在嬰兒型、線樣體出現在晚期嬰兒型指紋體出現在青少年型,青少年型患者存在不同形態的指紋體結構,這些特徵性的脂褐素顆粒需要在放大2萬倍才能觀察清楚,此外不要把正常的生理性脂褐素誤為病理性脂褐素。
5.免疫組織化學線粒體ATP合成酶C亞單位是CLN2、CLN3、CLN4CLN5CLN6和CLN8沉積物中的主要成分,免疫組織化學檢查證實這些蛋白的沉積有助於NCL的診斷。
臨床表現
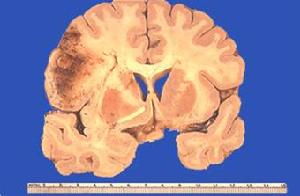 大腦白質
大腦白質本病具有隱性遺傳的特點偶爾在成年型NCL出現顯性遺傳,但僅有大約20%的患者有家族史14%的家族可以出現2個患病的兒童,一個家族出現3個和4個患者分別為3.2%和1.17%
80%NCL患者的首發症狀為癲癇,痴呆,失明或者運動障礙。20%的患者出現其他的首發症狀主要集中在青少年型的NCL,如行為異常、精神病周圍神經病、不隨意運動和共濟失調。出現非典型的NCL臨床表現可能是常見亞型的個體變異,如合併多發性周圍神經病,關節病和骨硬化病,有時很難區別是NCL的非典型表現還是兩個病的巧合。
1.嬰兒型CLN1發病年齡在0~2歲表現為幾乎完全的精神和運動功能衰竭,患者出現耐藥性癲癇發作和類似於脊髓休克的症狀,如:腱反射減低和肌張力低下無視網膜的累及症狀有的嬰兒表現為類似Rett綜合徵的臨床症狀,患者表現為智慧型和語言發育倒退不伴有癲癇和視網膜變性。
2.晚期嬰兒型NCL及其變異型
(1)經典的晚期嬰兒型NCL,CLN2:發病年齡在2~4歲半耐藥性癲癇和智力發育倒退為主要表現而後出現肌強直、共濟失調視力喪失和視神經萎縮,大部分患者在發病後3年半左右臥床不起,在10~15歲死亡除此之外這個亞型存在最多的變異型。Wisniewski變異型發病年齡在2歲半~3歲半首發症狀是由於小腦和錐體外系病變引起的運動異常,而後出現痴呆肌陣攣癲癇發作,視力障礙出現在5~6歲Edathodu變異型的發病年齡在9歲,患者主要表現為精神異常,不伴有癲癇痴呆運動異常和視網膜病變。
(2)芬蘭變異型NCL,CLN5:發病年齡在3~6歲,開始出現注意力不集中和運動的笨拙而後出現表現為智慧型發育遲緩、視力喪失、共濟失調肌陣攣和難治性癲癇。
(3)早期青少年型NCL,CLN6:即LINCL的非芬蘭變異型(Lake-Cavanagh病),屬於晚期嬰兒型的變異型,發病年齡在4~5歲,表現為共濟失調,而後出現視力喪失、癲癇發作和痴呆。非空泡性指紋體出現在血淋巴細胞中類似於青少年型的NCL。
3.青少年型,CLN3臨床表現也有明顯的差異典型患者的發病年齡在4~10歲,視力喪失和視網膜變性為主要表現,同時伴有癲癇和輕度的精神和智力損害。青少年型NCL的變異型首先表現為學習障礙,而後出現進行性的全腦性痴呆,失明,失語,最後在12~18歲出現不能進食和不能行走。延遲性青少年型表現為在10~20歲出現視力損害,繼而出現癲癇和痴呆。患者可以生存到40歲病理改變表現為線樣體和指紋樣體過去此型被作為成年型NCL進行了報導,但延遲性青少年型的遺傳和病理改變特點不同於成年型的NCL。
 神經元蠟樣脂褐質沉積症
神經元蠟樣脂褐質沉積症以精神分裂症起病的Kufs病不在少數,這些患者表現出思維混亂、情感淡漠偏執、幻覺、行為失常,抑鬱等,持續很長時間後才出現神經病學表現詳細的神經系統檢查顯然對疾病的診斷具有提示意義。
5.進行性癲癇伴智慧型發育延遲,CLN8此型是一種出現在芬蘭的東北部的NCL亞型也是常染色體隱性遺傳性疾病。此病早期正常發育有非常拖延的病程發病年齡在5~10歲,主要表現為癲癇大發作,而後出現進行性的智慧型發育延遲癲癇在青春期以前發作頻率增加而後發作減少痴呆出現在癲癇發作後2~5年,持續到成年部分患者出現構音障礙和行為異常視力的改變比較輕微或後期出現患者的壽命比其他NCL長。
併發症
隨著病情發展,不同臨床亞型的症狀體徵複雜多樣,可以是本病表現,也可以看作本病併發症(參見上述臨床表現)。特別是應注意合併的智慧型發育遲緩、痴呆、失明、失語,肺部感染、跌傷等。
診斷
 基因治療
基因治療診斷此病主要依靠臨床表現病理檢查結果和基因檢查結果。其中病理檢查發現病理性脂褐素顆粒是診斷NCL的金標準。
產前診斷NCL也主要依靠電子顯微鏡和基因技術。通過絨毛膜活檢,檢查基質血管壁幾乎可以100%地診斷嬰兒型NCL,在懷孕12周時50%的基質血管壁出現顆粒脂色素,如果檢查40個基質血管沒有發現脂色素的沉積基本可以除外嬰兒型NCL電子。顯微鏡檢查可以單獨套用於晚期嬰兒型NCL和其芬蘭變異型的產前診斷,曲線體可以在羊膜細胞中發現胎兒皮膚活檢也有利於診斷,晚期嬰兒型NCL的芬蘭變異型基因檢查發現CLN5的基因突變。但對青少年型NCL的電鏡檢查目前尚無一致的意見,其產前診斷主要依靠基因檢查。CNL1、CLN2CLN3和CLN5目前均可以根據基因的異常來進行產前診斷。
鑑別診斷
應注意與其他錐體外系病變引起的運動異常共濟失調,以及各類型痴呆精神異常延髓麻痹肌陣攣型癲癇和視網膜病變等鑑別。
檢查
實驗室檢查:本病一般實驗檢查,如血、尿、便腦脊液常規檢查大多數無明顯異常。
其它輔助檢查:
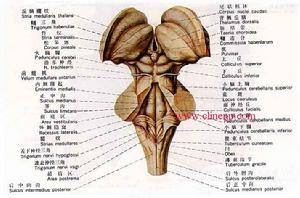 神經元蠟樣脂褐質沉積症
神經元蠟樣脂褐質沉積症1.影像學檢查MRI檢查對於NCL沒有特異性,但有助於NCL的鑑別診斷。NCL的MRI特點包括:
(1)瀰漫性腦萎縮:是主要的影像學改變,表現為腦室和腦溝擴大,在CLNl和CLN2表現比較明顯,特別是小腦萎縮在CLN3和CLN4一般在早期不明顯,在晚期主要表現為大腦和小腦的萎縮。
(2)大腦白質在T2相出現信號輕度增高:主要是深部大腦白質的改變,一般首先出現在側腦室后角附近的白質,後期出現胼胝體萎縮,腦幹和小腦白質無明顯改變改變的程度不如腦白質營養不良明顯。
(3)皮質變薄:出現的比較晚在橫斷面比較有助於觀察。
(4)丘腦在T2相低密度MRI的異常改變可以出現在亞臨床狀態,圖像改變隨病程的延長而加重,形態改變在前4年發展迅速在病程晚期腦萎縮更加顯著
SPECT顯示有廣泛的灰質葡萄糖代謝減少或缺乏這種改變以丘腦和皮質最為明顯並且和病情輕重及病程長短有明顯的相關性。
2.電生理檢查體感聽覺和視覺誘發電位異常以及視網膜電位的改變對於診斷具有較高的提示價值腦電圖除發現患者有癲癇的電生理改變外,在低頻光刺激時出現多相高壓尖波是一種比較典型的,電生理改變CLN2出現假周期型的癲癇放電在CLN4可以發現肌陣攣的改變特點。
3.形態學檢查屍體解剖是診斷此病最經典的可靠方法,超微結構檢查發現典型的病理性脂褐素是診斷NCL的金標準皮膚和血淋巴細胞的電鏡檢查是目前最常用的確診此病的手段,早期的腦活檢、器官活檢目前已經放棄肌肉活檢也較少被套用。應當注意,約15%的患者第一次皮膚活檢沒有陽性發現,同時結合進行血淋巴細胞檢查將有助於提高陽性率。病理性脂色素顆粒,主要出現在皮膚汗腺分泌部的上皮細胞,在皮膚平滑肌細胞、血管內皮和Schwann細胞較少見皮脂腺和大汗腺一般不受累及上皮細胞和纖維細胞也極少被累及,因此皮膚活檢必須取到汗腺的分泌部。成年型NCL脂色素也出現在中樞神經系統以外的軀體細胞內此外尿沉渣的腎小管上皮細胞檢查和尿ATP合成酶的C亞單位檢查也有助於晚期嬰兒型和青少年型NCL的診斷。
4.基因檢查基因檢查目前已經成為診斷NCL的重要方法,是除形態學檢查之外一個可靠的診斷手段。但在晚期嬰兒型由於存在許多變異型個別亞型的基因改變不清楚,中國患者是否存在和西方相同的NCL基因改變還不明確所以診斷價值還有待進一步提高。
治療
尚無有效治療,可給予抗癲癇藥物等對症處理有文獻報告早期採取骨髓移植的方法治療此病有一定的療效基因治療和神經幹細胞移植,可能是未來治療此病的研究方向。
預後:本病預後不良嬰兒型表現為幾乎完全的精神和運動功能衰竭,智慧型和語言發育倒退。晚期嬰兒型NCL及其變異型的大部分患者,在發病後3年半左右臥床不起,在10~15歲死亡青少年型NCL的變異型,首先表現為學習障礙而後出現進行性的全腦性痴呆,失明,失語,最後在12~18歲出現不能進食和不能行走進行性癲癇伴智慧型發育延遲發病年齡在5~10歲,主要表現為癲癇大發作,而後出現進行性的智慧型發育延遲持續到成年部分患者出現構音障礙和行為異常患者的壽命比其他NCL長。
預防:尚無有效的預防方法對症處理是臨床醫療護理的重要內容。
