基本定義
 實用臨床醫學1921年Hunt在題為“肌陣攣性小腦協調不能——原發性齒狀核萎縮”的文章中描述4例散發病例和1對孿生兄弟。這對孿生兄弟於兒童早期的表現類似Friedreich共濟失調,在30歲左右出現肌陣攣和癲癇,其中1例36歲死亡,屍檢顯示脊髓小腦束和後索變性,齒狀核萎縮,小腦上腳變細。因此,Hunt認為肌陣攣和小腦性共濟失調有特殊的關係,並推斷小腦齒狀核系統的原發性萎縮是肌陣攣和小腦性共濟失調的病理基礎,隨後,以Ramsay Hunt命名的許多相似病例也相繼被報導。然而,關於RHS是作為一個疾病的實體,還是一個綜合徵,一直是近期爭論的焦點,臨床上人們很難將其與進行性肌陣攣性癲癇(progressive myoclonic epilepsy,PME)中的一種類型——Unverrich-Lundborg病相區分,它們是否屬於同一種病尚不清楚。1990年,Marseille協作小組將RHS分成兩大類,即PME和進行性肌陣攣性共濟失調(progressive myoclonic ataxia, PMA)。PME是指肌陣攣伴有癲癇發作和進行性神經功能衰退,如輕度共濟失調和痴呆。PMA是指肌陣攣、進行性小腦性共濟失調,癲癇發作並不頻繁。
實用臨床醫學1921年Hunt在題為“肌陣攣性小腦協調不能——原發性齒狀核萎縮”的文章中描述4例散發病例和1對孿生兄弟。這對孿生兄弟於兒童早期的表現類似Friedreich共濟失調,在30歲左右出現肌陣攣和癲癇,其中1例36歲死亡,屍檢顯示脊髓小腦束和後索變性,齒狀核萎縮,小腦上腳變細。因此,Hunt認為肌陣攣和小腦性共濟失調有特殊的關係,並推斷小腦齒狀核系統的原發性萎縮是肌陣攣和小腦性共濟失調的病理基礎,隨後,以Ramsay Hunt命名的許多相似病例也相繼被報導。然而,關於RHS是作為一個疾病的實體,還是一個綜合徵,一直是近期爭論的焦點,臨床上人們很難將其與進行性肌陣攣性癲癇(progressive myoclonic epilepsy,PME)中的一種類型——Unverrich-Lundborg病相區分,它們是否屬於同一種病尚不清楚。1990年,Marseille協作小組將RHS分成兩大類,即PME和進行性肌陣攣性共濟失調(progressive myoclonic ataxia, PMA)。PME是指肌陣攣伴有癲癇發作和進行性神經功能衰退,如輕度共濟失調和痴呆。PMA是指肌陣攣、進行性小腦性共濟失調,癲癇發作並不頻繁。病理生理
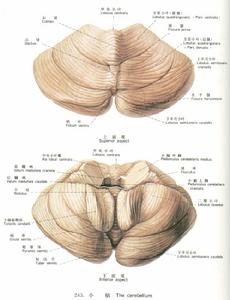 小腦齒狀核病因不明,與遺傳有關,Hunt對1例肌陣攣伴小腦性共濟失調病人進行屍檢時發現小腦齒狀核原發性萎縮,小腦上腳變細;其他小腦結構,皮層、橄欖小腦系統和橋腦小腦系統均完好,最早提出小腦齒狀核及傳出系統是肌陣攣和小腦性共濟失調的病理基礎。1994年,Kobayashi等的1例RHS屍檢病理報導顯示額葉白質脫髓鞘、皮層下纖維膠質細胞顯著增生,齒狀核和下橄欖核呈凝塊樣變性,骨骼肌沒有不整紅邊纖維(RRF),也認為肌陣攣和共濟失調與齒狀核的凝塊樣變性密切相關。新小腦和齒狀核傳出系統與保持肢體的姿勢,尤其是近端關節通過遠端手指操作來調整姿勢有關,因而可以說明肌陣攣為什麼發生於體位變化和運動時。肌陣攣是由於與運動有關的感覺(如聲音、視覺和軀體感覺)誘發運動的一種過激反應,正常人感覺傳入很快,經中樞神經系統處理後引起肌肉興奮,而小腦的核團是通過丘腦腹外側核與皮層聯繫來調節運動,齒狀核病變使感覺傳入到運動皮層興奮的時程縮短。
小腦齒狀核病因不明,與遺傳有關,Hunt對1例肌陣攣伴小腦性共濟失調病人進行屍檢時發現小腦齒狀核原發性萎縮,小腦上腳變細;其他小腦結構,皮層、橄欖小腦系統和橋腦小腦系統均完好,最早提出小腦齒狀核及傳出系統是肌陣攣和小腦性共濟失調的病理基礎。1994年,Kobayashi等的1例RHS屍檢病理報導顯示額葉白質脫髓鞘、皮層下纖維膠質細胞顯著增生,齒狀核和下橄欖核呈凝塊樣變性,骨骼肌沒有不整紅邊纖維(RRF),也認為肌陣攣和共濟失調與齒狀核的凝塊樣變性密切相關。新小腦和齒狀核傳出系統與保持肢體的姿勢,尤其是近端關節通過遠端手指操作來調整姿勢有關,因而可以說明肌陣攣為什麼發生於體位變化和運動時。肌陣攣是由於與運動有關的感覺(如聲音、視覺和軀體感覺)誘發運動的一種過激反應,正常人感覺傳入很快,經中樞神經系統處理後引起肌肉興奮,而小腦的核團是通過丘腦腹外側核與皮層聯繫來調節運動,齒狀核病變使感覺傳入到運動皮層興奮的時程縮短。 損害猴的齒狀核及其傳出通路時可得到由破壞病灶引起的共濟失調和意向性震顫,由刺激病灶引起的運動過度和肌陣攣,由於齒狀核中的各個區域可能與大腦皮層不同的運動區域有聯繫,因此齒狀核的不同核團受到刺激放電引起不同部位的肌陣攣,肌陣攣並不是齒狀核嚴重的急性損害所致,而是慢性刺激所致。
臨床表現
 腦部臨床RHS多在7~21歲發病,可發生於同一家族中,男女均可發病,為常染色體顯性遺傳外顯不全或常染色體隱性遺傳,故散發居多。突出表現為動作性肌陣攣、癲癇發作和小腦性共濟失調。肌陣攣表現為短暫的、突發的、無規律性的肌肉收縮,可以是全身的或局限於一組或多組肌群,無意識喪失,對運動、光、聲音刺激很敏感,體位改變、睡眠和情緒變化均可使其發作或加重,可能是與運動有關的感覺刺激的一種過激反應,偶爾可以出現全身強直陣攣性發作,但並不頻繁。肌陣攣常和小腦性共濟失調並存,表現為構音不清、意向性震顫、辨距不良等,共濟失調可先與肌陣攣或在肌陣攣數年後出現小腦症狀,早期可能由於小腦症狀較輕而被肌陣攣所掩蓋。多數無智慧型減退或輕度的認知障礙。本病進展緩慢,不影響壽命。
腦部臨床RHS多在7~21歲發病,可發生於同一家族中,男女均可發病,為常染色體顯性遺傳外顯不全或常染色體隱性遺傳,故散發居多。突出表現為動作性肌陣攣、癲癇發作和小腦性共濟失調。肌陣攣表現為短暫的、突發的、無規律性的肌肉收縮,可以是全身的或局限於一組或多組肌群,無意識喪失,對運動、光、聲音刺激很敏感,體位改變、睡眠和情緒變化均可使其發作或加重,可能是與運動有關的感覺刺激的一種過激反應,偶爾可以出現全身強直陣攣性發作,但並不頻繁。肌陣攣常和小腦性共濟失調並存,表現為構音不清、意向性震顫、辨距不良等,共濟失調可先與肌陣攣或在肌陣攣數年後出現小腦症狀,早期可能由於小腦症狀較輕而被肌陣攣所掩蓋。多數無智慧型減退或輕度的認知障礙。本病進展緩慢,不影響壽命。較常見的有:Unverricht-Lundborg病(亦稱波羅的海肌陣攣)、Lafora體病、神經元蠟樣褐脂質沉積症、線粒體腦肌病MERRF型、Sialidosis病(櫻桃紅斑肌陣攣綜合徵)。
較少見的有:Gaucher病、GM2神經節苷脂沉積症、生物素反應性腦病、運動性肌陣攣-腎臟損害綜合徵、齒狀核紅核-蒼白球丘腦底核萎縮、Ekbom綜合徵(PME、痴呆、脂肪瘤、骨骼畸形)、May-White綜合徵(PME、聽力喪失、共濟失調)等。
 臨床薈萃
臨床薈萃所有這些疾病都有獨特的生化及病理改變,亦具有不同的遺傳學特徵,如線粒體腦肌病MERRF型屬母系遺傳,由於線粒體的結構和功能異常所致,可伴有血及腦脊液乳酸和丙酮酸水平增高,肌活檢可見RRF纖維,目前已有人證實其基因突變的位點位於線粒體DNA的tRNALysA-G8344上。Unverricht-Lundborg病肌陣攣較突出,生化及病理無異常,而Lafora體病智慧型減退較明顯,肌陣攣較輕,腦電圖顯示特徵性枕葉癲癇波釋放,腦、肝、骨骼肌、皮膚活檢可見Lafora小體,神經元蠟樣脂褐質沉積症與脂肪酸過氧化酶缺乏有關,Sialidosis病的病因是α-N-乙醯神經胺酶和β-半乳糖苷酶缺乏,Gaucher病是由於葡萄糖腦苷酶缺乏等所致。由於上述疾病都可以引起肌陣攣和共濟失調,臨床上很難將RHS與上述疾病區分開來,特別是Unverricht-Lundborg的診斷標準(6~15歲起病,表現肌陣攣、強直-陣攣發作、特徵性腦電圖改變,呈進行性)與RHS基本相同。Marsden等研究了30例過去診斷為RHS的病人,對其中13例進行肌活檢,5例證實為線粒體腦肌病,而相當一部分人病因不明,其中4例符合Unverricht-Lundborg的診斷標準,故我們認為,RHS不能作為一個獨立的疾病實體存在,而是一個臨床綜合徵,可以由多種原因引起,臨床醫生也不能把RHS作為最後診斷,而應從多方面排出上述疾病。
生理改變
 癲癇兒做腦電圖腦電圖改變雖無特異性,但腦電圖的研究有助於揭示RHS的病變範圍和類型,腦電圖上可見到廣泛或散在的棘波、多棘波、多棘慢波暴發,多為雙側,可被光刺激誘發,RHS不同的腦電圖改變可能反映不同的病因。Bergamaso等發現,RHS病人的睡眠時快速眼動睡眠消失,說明睡眠周期被破壞,並伴全身性肌陣攣發作,腦電圖上快速眼動睡眠階段被陣發的棘波代替,經椎動脈注射異戊巴比妥可暫時抑制棘波,表明小腦和腦幹在疾病的發生和發展過程中起作用。Benassi等發現快速眼動睡眠、睡眠波、K-複合波和高尖波均減少。而Tassinari等發現10例RHS病人的結果卻與之相反,所有病人的睡眠周期保留完好。清醒時可見4~5周期/秒的棘波,肌陣攣發作時肌電圖可見肌陣攣波,同時腦電圖上也可見棘波。Kunesch等觀察發現,當RHS病人手指受到刺激時,在感覺運動皮層可記錄到巨大的皮層電位,從手指受到刺激至肌陣攣出現的潛伏期明顯延長。
癲癇兒做腦電圖腦電圖改變雖無特異性,但腦電圖的研究有助於揭示RHS的病變範圍和類型,腦電圖上可見到廣泛或散在的棘波、多棘波、多棘慢波暴發,多為雙側,可被光刺激誘發,RHS不同的腦電圖改變可能反映不同的病因。Bergamaso等發現,RHS病人的睡眠時快速眼動睡眠消失,說明睡眠周期被破壞,並伴全身性肌陣攣發作,腦電圖上快速眼動睡眠階段被陣發的棘波代替,經椎動脈注射異戊巴比妥可暫時抑制棘波,表明小腦和腦幹在疾病的發生和發展過程中起作用。Benassi等發現快速眼動睡眠、睡眠波、K-複合波和高尖波均減少。而Tassinari等發現10例RHS病人的結果卻與之相反,所有病人的睡眠周期保留完好。清醒時可見4~5周期/秒的棘波,肌陣攣發作時肌電圖可見肌陣攣波,同時腦電圖上也可見棘波。Kunesch等觀察發現,當RHS病人手指受到刺激時,在感覺運動皮層可記錄到巨大的皮層電位,從手指受到刺激至肌陣攣出現的潛伏期明顯延長。病症治療
 氯硝安定RHS診斷主要依靠臨床特徵,並且排除有關疾病。診斷要點:(1)7~21歲發病,有家族遺傳史,也可散發;(2)臨床特徵:肌陣攣、小腦性共濟失調,可伴有癲癇發作,但不頻繁;(3)腦電圖顯示特徵性的棘波、多棘波、棘慢波暴發。體感誘發電位可見巨大的皮層電位;(4)排除其他疾病:線粒體腦肌病、Sialidosis病、神經節蠟樣脂褐質沉積症、Lafore體病、Gaucher病等。
氯硝安定RHS診斷主要依靠臨床特徵,並且排除有關疾病。診斷要點:(1)7~21歲發病,有家族遺傳史,也可散發;(2)臨床特徵:肌陣攣、小腦性共濟失調,可伴有癲癇發作,但不頻繁;(3)腦電圖顯示特徵性的棘波、多棘波、棘慢波暴發。體感誘發電位可見巨大的皮層電位;(4)排除其他疾病:線粒體腦肌病、Sialidosis病、神經節蠟樣脂褐質沉積症、Lafore體病、Gaucher病等。 小腦變性目前尚無特殊療法,對肌陣攣及癲癇發作可以採用安定類或丙戊酸鈉等抗癲癇類藥物治療氯化安定劑量為7.5~15mg/d鵻分3次口服。氯硝安定效果最好,重者選用氯硝西泮(氯硝安定)成人口服1~2mg/次健康搜尋,3~4次/d,每天最大劑量為10mg。副作用有抑制呼吸、降低血壓及嗜睡等。丙戊酸鈉可用於同時有癲癇發作健康搜尋的患者鵻,成人劑量0.6~1.2g/d分3次口服。副作用為嗜睡、胃腸道反應及肝臟等損害對於小腦性共濟失調可採用康復訓練以延緩病情發展健康搜尋。聯合用藥較單一效果要好,如氯硝安定和丙戊酸鈉或撲癇酮等聯合用藥,嚴重病人甚至可以3種藥物聯合使用。對小腦性共濟失調尚無對症治療的措施,物理療法和康復訓練也很重要。本病進展緩慢,不影響壽命,病程可持續數10年之久。
神經系統遺傳病治療困難健康搜尋療效健康搜尋不滿意預防顯得更為重要健康搜尋。預防措施包括避免近親結婚鵻推行遺傳諮詢、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生。
問題展望
 腦部CT雖然對RHS的認識已有70多年的歷史,但是目前對本病的認識還比較膚淺,還存在一些問題值得進一步探討:(1)RHS這一名稱並未被普遍認可,其恰當的命名、分類及與其他臨床表現類似的疾病相鑑別上還存在許多問題;(2)肌陣攣性癲癇和小腦性共濟失調的關係及其解剖和病理生理學機制尚不清楚;(3)運動性肌陣攣的存在使人們很難評價小腦功能;(4)原發性癲癇和肌陣攣的病人多年後出現共濟失調,是否與抗癲癇藥有關;(5)區分肌陣攣和非癲癇性生理性肌陣攣從臨床和電生理上是十分困難的;(6)RHS與PME之間的關係很混亂,它們既不能完全等同,但又有重疊。
腦部CT雖然對RHS的認識已有70多年的歷史,但是目前對本病的認識還比較膚淺,還存在一些問題值得進一步探討:(1)RHS這一名稱並未被普遍認可,其恰當的命名、分類及與其他臨床表現類似的疾病相鑑別上還存在許多問題;(2)肌陣攣性癲癇和小腦性共濟失調的關係及其解剖和病理生理學機制尚不清楚;(3)運動性肌陣攣的存在使人們很難評價小腦功能;(4)原發性癲癇和肌陣攣的病人多年後出現共濟失調,是否與抗癲癇藥有關;(5)區分肌陣攣和非癲癇性生理性肌陣攣從臨床和電生理上是十分困難的;(6)RHS與PME之間的關係很混亂,它們既不能完全等同,但又有重疊。 相關詞條
