疾病歷史
 一位唐氏綜合症患者在使用電鑽機
一位唐氏綜合症患者在使用電鑽機1866年,英國醫生約翰·朗頓·唐在學會首次發表了這一病症。它最早叫“蒙古症”或者“蒙古痴呆症”,因為唐醫生髮現他的病人的面部比正常人較寬,眼睛小而上挑,看起來與蒙古人種有類同之處。在現今醫學界認為這種叫法不尊重,也無醫學實際意義,而不再普遍使用。由於各國患者的面容有相似的特徵,唐氏綜合症病人也被俗稱為“國際人”。
1959年,法國遺傳學家傑羅姆·勒瓊(Jerome LeJeune)發現唐氏綜合症是由人體的第21對染色體的三體變異造成的現象。這也是人類首次發現的染色體缺陷造成的疾病。
1961年,“唐氏綜合症”一詞由《柳葉刀醫學期刊》(The Lancet)的編輯首先使用。
疾病概述
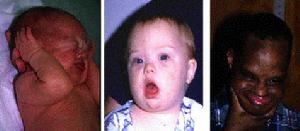 先天愚型
先天愚型疾病簡介
先天愚型又稱21三體綜合徵,三體綜合症或Down綜合症,屬常染色體畸變,是小兒染色體病中最常見的一種,活嬰中發生率約1/600~800,母親年齡愈大,本病的發病率愈高。60%患兒在胎兒早期即夭折流產。先天愚型包含一系列的遺傳病,其中最具代表性的第21對染色體的三體現象,會導致包括學習障礙、智慧型障礙和殘疾等高度畸形。這個病因在19世紀末,首次能描述它的病理的英國醫生唐·約翰·朗頓(John Langdon Down)而命名。
隨著產婦年齡的增長嬰兒患唐氏綜合徵的風險比率唐氏綜合徵患病機率高低與人種,生活水平等沒有直接聯繫,估計每660個新生兒就有一個患有唐氏綜合徵,使之成為最常見的染色體變異。高齡初產婦會加劇嬰兒患有唐氏綜合徵的風險。在20到24歲之間,患病率為1/1490,到40歲為1/106,49歲為1/11。原因是隨著產婦年齡的增加卵子形成過程中會引起染色體不分離現象的增加。但是,另一方面,大約80%的綜合徵患嬰是35歲以下產婦所生。這與35歲以下婦女妊娠比例較高有關係。
 唐氏綜合徵患者的瞳孔上會有典型的白點,叫Brushfield斑點
唐氏綜合徵患者的瞳孔上會有典型的白點,叫Brushfield斑點另外也有多餘的染色體來自父親一方的情況,父方起因和母方起因的比例為1:4。患病的潛在高風險家庭通常會被提議進行遺傳學諮詢和遺傳測試例如“羊水診斷”等。
唐氏綜合徵的小孩與不患病的小孩相比處於直接劣勢。通常認為患病兒童的精神發育遲緩,智商很少超過60的,腦部通常過小、過輕。小腦、腦幹和腦前回出奇地小。教育進度會因疾病和殘疾而遭到破壞,例如不斷復發的傳染病、心臟病、弱視、弱聽等。其他因患病造成的生理特徵包括猿皺等扭曲的面部特徵,有的唐氏綜合徵患者青年期以後由於壓力會引發抑鬱症。但總體上顯示出性格開朗的傾向。40歲以後患阿茲海默病比率高,容易出現記憶喪失、知能持續低下、人格變化等症狀。
身體特徵
以下特徵並非全部出現在患者身上,根據個人差異,也有身體特徵上不明顯的例子。 身材矮小,肌肉緊張度低下,體力低下,頸椎脆弱。頭部長度較常人短,面部起伏較小,鼻子,眼睛之間的部分較低,眼角上挑,深雙眼皮。耳朵上方朝內側彎曲,耳朵整體看上去呈圓形而且位置較低。舌頭比較大。脖子粗壯。手比較寬,手指較短,拇指和食指之間間隔較遠,小指缺少一個關節,向內彎曲。手掌的橫向紋路只有一條,指紋為弓狀。腳趾第一趾與第二趾之間間隔也比較大。
醫學研究
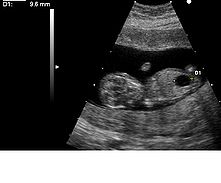 超音波顯示出一個患上唐氏綜合徵與巨膀胱的胎兒。
超音波顯示出一個患上唐氏綜合徵與巨膀胱的胎兒。正常染色體按照大小順序編號由1到22號,染色體早期的檢查中,曾誤認為唐氏綜合徵患者較大的染色體是第21對,其後的研究表明是源於較小的染色體的異常。但是為了不引起混亂,將第21對與第22對的名稱對調,現在繼續沿用“第21對染色體三體變異”這一名稱。
唐氏綜合徵患者的第21對染色體多出一條變成三條。第21對染色體是最小的染色體,其攜帶的遺傳信息也相對較少,比較其他染色體的三體變異現象屬於輕度先天異常。所以能將患病嬰兒正常產下的幾率較高。
有關嬰兒出生前可導致智力問題的疾病,以唐氏綜合徵的研究最多及最詳盡。根據研究個案顯示,95%的患者的第21對染色體都有異常(變成有3條)的現象。而餘下的5%,都是因為在第21對染色體的相關部份重複了,比如各種第21對染色體的位置轉移。照病理學的臨床研究,患者的智力障礙從中度到重度的都有。
疾病病因
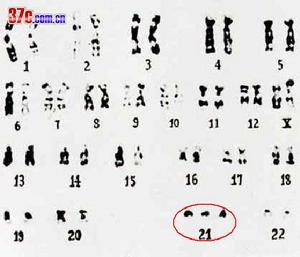 先天愚型 病因
先天愚型 病因先天愚型是第21對染色體的三體變異現象。 染色體移位造成第14對染色體的變異標準型第21對染色體三體變異(Trisomy 21):又稱21三體綜合症,第21對染色體多出一條,細胞中有四十七條色體,占唐氏綜合症患者的90-95% 這大多是由通常第1減數分裂期的不分離造成的。也有在第2減數分裂時發生的情況。父母方通常都攜帶正常的染色體,嬰兒是偶然形成的三體異常。染色體移位型(Translocation):占全體比例的5-6% 。
細胞中多出一條染色體,附著在D組(第13、14、15對染色體)或者G組(第21對22對染色體)的染色體上,特別容易出現在第14對或第21對染色體上。移位型中一半左右是偶發性的,也就是父母雙方都是正常染色體。另外一半是遺傳性移位,父母有一方攜帶有這樣的染色體,這在家族中常能找到相同病症的親屬。
無色體型(Mosaicism):占全體比例的1-3% .第21對三體變異染色體結合體(占80%)和正常細胞結合體的體細胞分裂所產生的不分離造成。這種場合表現出來的臨床現象較輕。通常父母方染色體正常,染色體的不分離在受精卵的細胞分裂過程中偶然發生,造成嬰兒的部分細胞三體變異,部分細胞正常,極為罕見。
發病機制
 先天愚型
先天愚型先天愚型患病幾率高低與人種,生活水準等沒有直接聯繫,估計每660個新生兒就有一個患有唐氏綜合症,使之成為最常見的染色體變異。高齡初產婦會加劇嬰兒患有唐氏綜合症的風險。在20到24歲之間,患病率為1/1490,到40歲為1/106,49歲為1/11。原因是隨著產婦年齡的增加卵子形成過程中會引起染色體不分離現象的増加。但是,另一方面,大約80%的綜合症患嬰是35歲以下產婦所生。這與35歲以下婦女妊娠比例較高有關係。 另外也有多餘的染色體來自父親一方的情況,父方起因和母方起因的比例為1:4。患病的潛在高風險家庭通常會被提議進行遺傳學諮詢和遺傳測試例如“羊水診斷”等。
隨著產婦年齡的增長嬰兒患先天愚型的風險比率先天愚型的小孩與不患病的小孩相比處於直接劣勢。通常認為患病兒童的精神發育遲緩,智商很少超過60的,腦部通常過小、過輕。小腦、腦幹和腦前回出奇地小。教育進度會因疾病和殘疾而遭到破壞,例如不斷復發的傳染病、心臟病、弱視、弱聽等。其他因患病造成的生理特徵包括猿皺等扭曲的面部特徵,有的先天愚型患者青年期以後由於壓力會引發抑鬱症。但總體上顯示出性格開朗的傾向。40歲以後患阿茲海默病比率高,容易出現記憶喪失、知能持續低下、人格變化等症狀。
細胞遺傳學
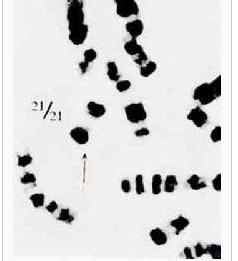 21三體綜合徵又稱先天愚型
21三體綜合徵又稱先天愚型按照核型分析可將先天愚型患兒分為三型,其中標準型和易位型在臨床上不易區別,嵌合型的臨床表現差異懸殊,視正常細胞株所占的百分比而定,可以從接近正常到典型表型。
(一)標準型
患兒體細胞染色體為47條,有一條額外的21號染色體,核型為47。XX(或XY),+21,此型占全部病例的95%。其發生機制系因親代(多數為母方)的生殖細胞染色體在減數分裂時不分離所致。雙親外周血淋巴細胞核型都正常。
(二)易位型
約占2.5%~5%。多為羅伯遜易位(Robertsonian translocation),是只發生在近端著絲粒染色體的一種相互易位,亦稱著絲粒融合,其額外的21號染色體長臂易位到另一近端著絲粒染色體上。其中,D/G易位最常見,D組中以14號染色體為主,即核型為 46,XX(或 XY)-14,+t(14q21q);少數為15號。這種易位型患兒約半數為遺傳性,即親代中有 14/21平衡易位染色體攜帶者,核型為 45,XX(或 XY),-14,-21,+t(14q21q)。另一種為G/G易位,是由於G組中兩個21號染色體發生著絲粒融合,形成等
臂染色體t(21q21q),或一個21號易位到一個22號染色體上,t(21q22q),較少見。
(三)嵌合體型
約占本症的2%~4% 患兒體內有兩種以上細胞株(以兩種為多見),一株正常,另一株為21-三體細胞,本型是因受精卵在早期分裂過程中染色體不分離所引起,臨床表現隨正常細胞所占百分比而定。
疾病種類
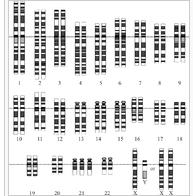 第21對染色體的三體變異現象
第21對染色體的三體變異現象先天愚型基本上分為三種類型:
標準型第21對染色體三體變異(Trisomy 21):又稱廿一三體症,第21對染色體多出一條,細胞中有四十七條色體,占唐氏綜合徵患者的90-95%
這大多是由通常第1減數分裂期的不分離造成的。也有在第2減數分裂時發生的情況。父母方通常都攜帶正常的染色體,嬰兒是偶然形成的三體異常。
染色體移位型(Translocation):占全體比例的5-6%
細胞中多出一條染色體,附著在D組(第13、14、15對染色體)或者G組(第21對22對染色體)的染色體上,特別容易出現在第14對或第21對染色體上。移位型中一半左右是偶發性的,也就是父母雙方都是正常染色體。另外一半是遺傳性移位,父母有一方攜帶有這樣的染色體,這在家族中常能找到相同病症的親屬。
無色體型(Mosaicism):占全體比例的1-3%
由第21對三體變異染色體結合體(占80%)和正常細胞結合體的體細胞分裂所產生的不分離造成。這種場合表現出來的臨床現象較輕。通常父母方染色體正常,染色體的不分離在受精卵的細胞分裂過程中偶然發生,造成嬰兒的部分細胞三體變異,部分細胞正常,極為罕見。
危險因素
先天愚型是一種偶發性疾病,所以每一個懷孕的婦女都有可能生出“唐氏兒”。生唐氏兒的幾率會隨著孕婦年齡的遞增而升高。下列7類夫妻屬高發人群:
1、妊娠前後,孕婦有病毒感染史,如流感、風疹等;
2、受孕時,夫妻一方染色體異常;
3、夫妻一方年齡較大;
4、妊娠前後,孕婦服用致畸藥物,如四環素等;
5、夫妻一方長期在放射性熒幕下工作或污染環境下工作;
6、有習慣性流產史、早產或死胎的孕婦;
7、夫妻一方長期飼養寵物者。
八大特徵
 唐氏綜合症
唐氏綜合症2、語言發育障礙:患兒開始學說話的平均年齡為4-6歲,95%有發音缺陷、口齒含糊不清、口吃、聲音低啞;1/3以上有語音節律不正常,甚至呈爆發音。
3、行為障礙:大多性情溫和,常傻笑,喜歡模仿和重複一些簡單的動作,可進行簡單的勞動,少數患者易激惹、任性、多動,甚至有破壞攻擊行為,某些則顯示畏縮傾向,伴有緊張症的姿勢。
4、運動發育遲緩:患兒在出生後的一段時期其運動功能與正常同齡兒差別可能不大,但隨年齡增長其差別增大。在不同的患者中運動發育的情況也相差很大。先天愚型患者可執行簡單的運動,如穿衣、吃飯等,但動作笨拙、不協調、步態不穩。
5、生長發育障礙:先天愚型患者母體妊娠期較短,平均為262~272天。出生時身高較正常新生兒短l~3cm,頭圍基本正常,雙頂徑在正常範圍,前後徑相對較短,枕部平坦。大多數呈短頭畸型。前後囟及前額縫寬,閉合遲,常出現第三囟(後囟上方的矢狀縫增寬)。本病患兒出生後幾天睡眠較深,吸吮、吞咽十分緩慢,甚至完全不能,故 弄醒和餵養十分困難。80%的患兒肌張力普遍低下。
6、特殊的外貌:雙眼距寬,兩眼外角上斜,內眥贅皮,耳位低,鼻樑低,舌體寬厚,口常半張或舌伸出口外,舌面溝裂深而多,手掌厚而指短粗,末指短小常向內彎曲或有兩指節,40%患兒有通貫掌。跖紋中,拇趾球區脛側弓狀紋,拇趾與第二趾指間距大,關節韌帶鬆弛或見肌張力低。
7、約有1/2的病例並發先天性心臟病、易患傳染性疾病和白血病。8.指紋改變
併發症
 先天愚型併發症 先心病
先天愚型併發症 先心病先天愚型患者伴隨有器髒畸形變異的幾率也較高,但並非所有併發症都會出現,也有完全沒有併發症的例子。
消化器官畸形,如先天性食道閉鎖症,十二指腸狹窄,鎖肛等。
先天性心臟病,患病比率高達40%,
尤其是心內膜不全比例較高,通常如果不進行早期治療會有致命危險。
白內障,患病率為2% ,急性白血病,患病率為1% 。
環軸間接不穩定性,患病率2-3% ,甲狀腺疾病,患病率3% ,點頭癲癇,患病率10%,一時性骨髓異常增生症,眼異常,由角膜,水晶體異常引發近視,遠視,亂視等 ,浸出性中耳炎,容易在內耳積蓄液體引發耳炎,影響聽覺。
臨床表現
先天愚型患兒的主要特徵為智慧型低下、體格發育遲緩和特殊面容。患兒眼距寬,鼻樑低平,眼裂小,眼外側上斜,有內毗贅皮,外耳小,硬齶窄小,舌常伸出口外,流涎多;身材矮小,頭圍小於正常,骨齡常落後於年齡,出牙延遲且常錯位;頭髮細軟而較少;四肢短,由於韌帶鬆弛,關節可過度彎曲,手指粗短,小指向內彎曲。
皮膚紋理特徵有:通貫手,aid角增大;第4,5指箕撓增多;腳拇指球脛側弓形紋和第5指只有一條指褶紋等。
患兒在出生時即已有明顯的特殊面容,且常呈現嗜題和餵養困難。隨著年齡增長,其智慧型低下表現逐漸明顯,動作發育和性發育部延遲。約30%患兒伴有先天性心臟病等其他畸形。因免疫功能低下,易患各種感染,白血病的發生率也增高10~30倍。如存活至成人期,則常在30歲以後出現老年性痴呆症狀。
疾病檢查
在妊娠11~13+6周,測量胎兒頸項透明層厚度(NT檢查)是篩查唐氏綜合徵等染色體異常的敏感指標。62%~80%的先天愚型胎兒可表現出NT增厚(大於3mm)。在妊娠14~16周左右,對羊水進行染色體檢查可以明確判明患病與否。這種檢查在一般的婦產科醫院就能進行。
但是根據各國相關規定不同,針對這種檢查的具體措施也有不同,比如在日本,基於一般的學會倫理規定,對這種出生前檢查不做積極的推薦,只有在妊婦自行主張,如結婚的夫婦必須徵得雙方同意的情況下,醫院才給與實施檢查。檢查結果也是在確認妊婦希望得到通知後才告之。
但是另一方面,如英國出生前診斷作為一項國家政策,這種檢查十分普及。
1.對外周血細胞染色體核型分析:細胞遺傳學研究發現,在21號染色體長臂21q22區帶為三體時,該個體具有完全類似唐氏綜合徵的臨床表現,相反,該區帶為非三體的個體則無此典型症狀。由此推論,21q22區可能是唐氏綜合徵的基因關鍵區帶,又稱為唐氏綜合徵區。按染色體核型分析可將唐氏綜合徵患兒分為3型:
(1)標準型:占全部病例的95%。患兒體細胞染色體為47條,有一個額外的21號染色體,核型為47,XX(或XY),+21。
(2)易位型:約占2.5%~5%,患兒的染色體總數為46條,多為羅伯遜易位(Robertsoniantranslocation),是指發生在近端著絲粒染色體的一種相互易位,多為D/G易位,D組中以14號染色體為主,即核型為46,XX(或XY),-14,+t(14q21q);少數為15號染色體易位,這種易位型患兒約半數為遺傳性,即親代中有平衡易位染色體攜帶者。另一種為G/G易位,較少見,是由於G組中2個21號染色體發生著絲粒融合,形成等臂染色體t(21q21q),或1個21號易位到1個22號染色體上。
(3)嵌合體型:約占本症的2%~4%,患兒體內有2種或者2種以上細胞株(以2種為多見),一株正常,另一株為21-三體細胞,臨床表現的嚴重程度與正常細胞所占百分比有關,可以從接近正常到典型表型,21-三體細胞株比例越高,智力落後及畸形的程度越重。
2.羊水細胞染色體檢查:羊水細胞染色體檢查是唐氏綜合徵產前診斷的一種有效方法,唐氏篩查結果為“高危”的孕婦需要確診胎兒是否為唐氏綜合症患兒。目前產前診斷最常用的技術是羊膜腔穿刺技術,即在B超指引下,將針通過孕婦腹部刺入羊水中,抽取羊水,對胎兒細胞進行染色體分析。適宜孕16~20周的孕婦。除羊膜腔穿刺術外,進行產前診斷的技術還有絨毛活檢、胎兒臍靜脈穿刺、胎兒鏡檢查等。常見核型與外周血細胞染色體核型相同。
3.螢光原位雜交:以21號染色體的相應部位序列作探針,與外周血中的淋巴細胞或羊水細胞進行雜交,唐氏綜合徵患者的細胞中可呈現3個21號染色體的螢光信號。若選擇唐氏綜合徵核心區的特異序列作為探針,進行FISH雜交分析,可以對21號染色體的異常部位進行精確定位,提高檢測21號染色體數目和結構異常的精確性。
4.產前篩查血清標誌物:採用測定孕婦血清絨毛膜促性腺激素(HCG)、甲胎蛋白(AFP)、游離雌三醇(FE3)進行唐氏綜合徵篩查的探索已有多年,這是一種懷孕中期指標(13周以後),根據這3項(也可測定HCG和AFP兩項)血清學指標以及孕婦年齡、體重來推算懷有唐氏綜合徵患兒的風險率,根據風險率的高低再進一步進行確診檢查。採用這一方法可以檢出大約60%的唐氏綜合徵胎兒。近年發現的妊娠相關血漿蛋白A(PAPP-A)的診斷價值日益受到重視。PAPP-A為胎盤滋養層細胞產生,早期懷唐氏綜合徵胎兒的孕婦血清水平明顯降低,推測可能與滋養層細胞功能降低有關。這是一種懷孕早期篩查指標,適用於懷孕8~14周孕婦篩查。採用PAPP-A、HCG、AFP等指標篩查不僅可篩查出唐氏綜合徵,還可檢出18-三體綜合徵、先天性神經管缺陷、先天性腹壁缺損等其他先天異常。此外,通過B超測量胎兒頸項皮膚厚度也是診斷唐氏綜合徵的重要指標。因此,對懷孕早、中期的孕婦開展唐氏綜合徵篩查,及早採取積極預防措施,對保證婦幼健康水平有一定意義。
5.可常規做X線片、超聲、心電圖、腦電圖等檢查,部分患兒可發現先天性心臟病,骨齡落後,腦電圖異常等改變。
治療護理
嬰兒
沒有相應的治療方法。在循環器併發症無法進行外科治療的幾十年前患者平均壽命只有20歲左右,現在如果對其並發的畸形進行治療能保持患者的身體健康狀態,而且平均壽命已經增加到50歲左右。甚至有的患者還通過努力完成了四年制大學學業。另外,早期的養育護理有助於患者的發育。
母親
通常生下患有唐氏綜合徵嬰兒對母親是很大的打擊,所以針對患者母親的精神護理尤為重要。
最新療法
幹細胞治療
幹細胞是一種未充分分化,尚不成熟的細胞,具有再生各種組織器官和人體的潛在功能,醫學界稱為“萬用細胞”。幹細胞治療是把健康的幹細胞移植或經靜脈注射到病人體內,以達到修復病變細胞或重建功能正常的細胞和組織為目的。幹細胞治療就像給機體注入新的活力,從根本上治療各種疑難疾病,也是目前治療唐氏綜合症唯一有效的方法。幹細胞治療疾病的基本原理:
1.對組織細胞損傷的修復。
2.替代損傷細胞的功能。
3.刺激機體自身細胞的再生功能。
 幹細胞治療
幹細胞治療社會爭議
檢查
出生前檢查現在受到廣泛的爭議。通常從醫學角度上講“針對35歲以上高齡妊婦的出生前檢查能高比例的發現病症”,但是這樣行為構成對唐氏綜合症患者的歧視,以及人權倫理上的問題,所以受到患者以及人權組織等強烈的反對。但是另一方面,一個家庭內如果有兩個患者會構成極大的負擔,一般為了防止第二胎同樣患病父母通常都會進行出生前檢查。
在不強烈反對墮胎的社會,會出現另一種爭議:大規模推廣高齡產婦的檢查後,唐式症新生兒數量只有稍微減少,因為有許多低年齡的產婦並沒有被提醒要做檢查;因此對於各年齡的產婦都應該提醒有生出唐式症嬰兒的風險。
墮胎
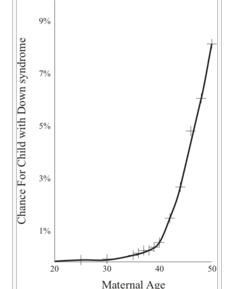 隨著產婦年齡的增長嬰兒患唐氏綜合徵的風險比率
隨著產婦年齡的增長嬰兒患唐氏綜合徵的風險比率即使出生前檢查發現胎兒患有唐氏綜合症,根據各國法律的不同規定,關於墮胎的措施也不相同。
在中國大陸,如果檢查出嬰兒患有唐氏綜合症,可以依照父母的意願決定墮胎與否。
日本根據《母體保護法》規定,不認可由於胎兒的問題而進行的人工墮胎。但是相關條文又說明“繼續妊娠或分娩會在身體或經濟理由上對母體健康造成顯著的傷害”或者“對母體來說構成極大的精神問題”或者“由於精神問題對母體構成了極大的健康障礙”的情況下,妊娠未滿22周前可以人工墮胎。但是墮胎的必要性需要指定醫生的確認,能夠進行的醫療機構也有指定。
流行文化
2008年7月20日,一位名叫希嘉·柏克絲(Helga Parks)的美國人,當她在許多年前於德國探親時,患有這個病症的侄女便送了一個像她自己的洋娃;加上在該國經常利用這類公仔教導學童不要對殘疾人士存有偏見,於是她便看準這個商機,回國後便設立貿易公司;開始進口這類公仔。結果當它推出市場後,各界人士褒貶不一;有人認為可以加深對此類病人的認識,不過亦有人認為只是標其立異。
早期干預
21三體綜合徵患兒並非不可矯正,通過早期干預,可以和正常人一樣學習生活和工作。
目前在中國普遍認為 21三體綜合徵患兒就是傻子,因為中國的唐氏患兒數量龐大,中國政府無法對其進行資助。在歐美國家,21三體綜合徵患兒基本均可正常生活。日本是掌握早期干預最成熟的國家,因為其對墮胎的要求十分嚴格。
