簡介
 患有甲基丙二酸尿症的雙胞胎嬰兒
患有甲基丙二酸尿症的雙胞胎嬰兒甲基丙二酸尿症(methylmalonicaciduria)或甲基丙二酸血症(methylmalonicacidemia)是先天有機酸代謝異常中最常見的病種,是多種原因所致體內甲基丙二酸蓄積的總稱,於1967年首次被報導。近年來,其病因、診斷、治療與遺傳學機制逐步明確,隨著質譜分析技術的普及,本症的流行病學資料亦不斷更新。據報導日本發病率為2/9780,2/102200至2/16246,義大利1/61775,中國發病情況不詳。
甲基丙二酸尿症是一種常染色體隱性遺傳代謝性疾病,表現為生長發育不良,智力落後或倒退,新生兒期發病嘔吐、脫水、厭食、呼吸急促甚至昏迷、死亡,急性期治療以補液、糾正酸中毒為主,必要時需透析。目前尚無特效療法,患者需要長期的營養治療和藥物治療。
病因分析
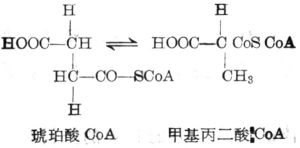 甲基丙二酸與琥珀酸的異構作用
甲基丙二酸與琥珀酸的異構作用甲基丙二酸尿症病因複雜。遺傳性甲基丙二酸尿症包括甲基丙二醯輔酶A變位酶酶蛋白(mutaseapoenzyme,mut)缺陷及其輔酶鈷胺素(維生素B12)代謝缺陷,均為常染色體隱性遺傳。甲基丙二醯輔酶A變位酶編碼基因位於6p21,迄今已發現10種突變,以導致胺基酸互換的錯義突變為多見。變位酶完全缺陷為mut0型,部分缺陷為mut-型。鈷胺素代謝障礙包括5類:兩種為腺苷鈷胺素(AdoCbl)合成缺陷,即線粒體鈷胺素還原酶(mito2chondrialCblreductase,CblA)缺乏,其基因定位為4q31.21和線粒體鈷胺素腺苷轉移酶(mitochodrialcobalaminadenosyltransferase,CblB)缺乏,其基因定位為12q24,3種為胞漿和溶酶體鈷胺素代謝異常所致腺苷鈷胺素和甲基鈷胺素(MeCbl)合成缺陷(CblC,CblD,CblF)。mut0,mut-,CblA和CblB型患者臨床表現類似,僅有甲基丙二酸尿症。CblC,CblD,CblF型患者生化特點為甲基丙二酸尿症合併同型半胱氨酸血症。根據患者對維生素B12的治療反應,臨床可分為維生素B12反應型和不反應型。維生素B12反應型患者多為輔酶合成缺陷,Cb1A,CblC,CblD,CblF型多為維生素B12反應型,Cb1B型中半數患者維生素B12有效。而維生素B12不反應型多為變位酶缺陷。
除上述遺傳缺陷外,轉鈷胺素Ⅱ缺陷、慢性胃腸與肝膽疾病、長期素食、特殊藥物治療可導致維生素B12缺乏,引起甲基丙二酸尿症。母親長期維生素B12攝入不足造成胎兒維生素B12缺乏,不僅引起母親惡性貧血,亦將導致嬰兒繼發性甲基丙二酸尿症,造血功能障礙,神經系統發育異常。
曾有研究調查了173例新生兒尿甲基丙二酸及其母親的血漿維生素B12及同型半胱氨酸濃度,證明出生6周內新生兒尿甲基丙二酸濃度與母親的血維生素B12水平呈負相關,與母親血液同型半胱氨酸濃度呈正相關。
發病機制
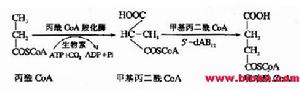 甲基丙二醯CoA變位酶的輔酶
甲基丙二醯CoA變位酶的輔酶甲基丙二酸是甲基丙二醯輔酶A的代謝產物,正常情況下在甲基丙二醯輔酶A變位酶及維生素B12的作用下轉化生成琥珀酸,參與三羧酸循環。甲基丙二醯輔酶A變位酶缺陷或維生素B12代謝障礙導致甲基丙二酸、丙酸、甲基枸櫞酸等代謝物異常蓄積,琥珀酸脫氫酶活性下降,線粒體能量合成障礙,引起神經、肝臟、腎臟、骨髓等多臟器損傷。患者腦組織病理分析可見腦萎縮、瀰漫性神經膠質細胞增生、星形細胞變性、腦出血、蒼白球壞死、丘腦及內囊細胞水腫,均與線粒體功能不良有關。
曾有病理解剖發現患兒神經膠質細胞反應性增生,深部皮質、小腦顆粒層和膠質細胞發育不良,小腦、腦幹、頸髓髓鞘化延遲。另有屍檢發現腎臟、肺部血栓性毛細血管病、肝臟瀰漫性脂肪變性、骨髓巨幼紅細胞增生、嚴重胃黏膜發育不良伴胃炎。這些表現部分為胎兒時期代謝異常所致損害,部分為出生後有機酸毒性損害所致。
臨床表現
 甲基鈷胺
甲基鈷胺起病
mut0型患者起病最早,80%在生後數小時至1周內發病,類似急性腦病樣症狀,如:拒乳、嘔吐、脫水、昏迷、驚厥、酸中毒、酮尿、低血糖,早期死亡率極高,預後不良。mut-及Cb1A和Cb1B型患者多在生後1月後發病,Cb1C和Cb1D在新生兒期至成年發病者均有報導,Cb1F報導很少。
發作誘因
部分患者呈急性發病或間歇性發病,發熱、感染、飢餓、疲勞、外傷等應激狀態下機體能量需求增加,高蛋白飲食、輸血等因素引起甲基丙二酸前身物質蛋氨酸、蘇氨酸、異亮氨酸、纈氨酸蓄積,丙戊酸、大環內酯類藥物導致左鏇肉鹼消耗,甲基丙二酸排泄障礙,引起急性代謝紊亂。
一般表現
患者臨床表現常無特異性,常常被誤診為一般圍產期腦損害、敗血症、急慢性腦病或腦變性病,常見餵養困難、嘔吐、呼吸急促、驚厥、肌張力異常、嗜睡、智力、運動落後或倒退,急性期可見昏迷、呼吸暫停、代謝性酸中毒、酮症、低血糖、高乳酸血症、高氨血症、高甘氨酸血症、肝損害、腎損害,嚴重時腦水腫、腦出血。患者神經系統異常各不相同,多於嬰兒期出現智力、運動落後,肌張力低下,變位酶缺陷患者常較鈷胺素代謝異常患者神經系統損害出現早,並且嚴重。少數鈷胺素代謝異常所致良性甲基丙二酸尿症患者可於成年後發病,甚至終身不發病。嚴重患者由於免疫功能低下,易合併皮膚念珠菌感染,常見口角、眼角、會陰部皸裂和紅斑。隨著
代謝紊亂的控制,患者皮膚損害逐漸恢復。
 臨床診斷
臨床診斷其它表現
甲基丙二酸尿症常導致多臟器損害。患者肝臟常明顯腫大,肝功能異常。骨質疏鬆較為常見,嚴重時可導致骨折。腎小管酸中毒、間質性腎炎、高尿酸血症、尿酸鹽腎病、遺尿症等慢性腎損害也屢見報導。嚴重患者合併溶血尿毒綜合徵,表現為微血管性溶血性貧血、血小板減少、腎功能衰竭、高血壓。患者血液系統異常多見巨幼細胞性貧血、粒細胞減少,血小板減少,嚴重時甚至出現骨髓抑制。少數患者合併口炎、舌炎、角膜潰瘍、一過性糖尿病等異常。甲基丙二酸尿症嬰兒常表現為相似面容,如:高前額、寬鼻樑、內眥贅皮、三角形嘴。患者個體差異很大,即使相同缺陷的同胞亦輕重不同。
影像學異常
甲基丙二酸尿症患者腦CT、MRI掃描常見對稱性基底節損害,以蒼白球損害為主。嬰幼兒患者白質發育落後、年長兒腦白質變性出現較早,隨病情進展出現瀰漫性腦萎縮。患者生後第1個月腦影像學可正常或白質變薄,1歲左右可發展為中到重度腦萎縮及白質發育落後。典型患者可見瀰漫性幕上白質水腫和髓鞘化不良,亦可因瀰漫性的顱內血管硬化導致腦積水。白質損傷可能與甲基化功能不良、非生理性的脂肪酸毒性有關。神經膠質細胞的增生與高半胱氨酸對內皮的毒性有關。
臨床診斷
由於個體差異較大,臨床誤診率較高,對於原因不明的嘔吐、驚厥、酸中毒、肌張力異常、發育落後、呼吸困難等患兒應及早進行有關檢查,尿酮體測定、血氣分析、血氨、血糖、心肌酶譜等一般生化檢查均有助於診斷。氣相色譜2質譜聯用分析尿、血、腦脊液有機酸定量檢測是確診本症的關鍵方法。正常人尿甲基丙二酸濃度<2mmol/mol·肌酐,24h的排出量<5mg,在甲基丙二醯輔酶A變位酶缺陷的患者尿甲基丙二酸濃度為270~13000mmol/mol·肌酐,24h總量可達240~5700mg。由於丙酸蓄積,患者尿32羥基丙酸和甲基枸櫞酸濃度亦明顯增高。經治療後患者尿甲基丙二酸排出可明顯減少。正常人血漿難以檢出甲基丙二酸,患者可達200~2500mmol/L。腦脊液甲基丙二酸濃度與血漿濃度接近。應注意排除新生兒期其它原因引起的酮症酸中毒、鈷胺素缺乏和其他原因所致同型半胱氨酸血症。
本症的病型分析需依賴皮膚成纖維細胞、淋巴細胞、肝組織纖維母細胞酶學分析或基因診斷。通過胎盤絨毛細胞、羊水細胞酶學檢查、母親尿液及羊水甲基丙二酸測定,可進行本症的產前診斷,國內外取得了成功的經驗。曾有患兒經產前檢查證實為脫氧腺苷鈷胺素合成酶缺陷,在出生前接受鈷胺素治療,得到了有效的控制。
治療方法
 臨床治療
臨床治療本症急性期的治療應以補液、糾正酸中毒為主,必要時進行腹腔透析或血液透析,同時,應保證熱量及液體供給以減少機體蛋白分解,必要時給予小量胰島素,限制天然蛋白質的攝入。鑒於重症患兒或代謝性酸中毒急性發作期死亡率極高,臨床高度懷疑時,可在確診前進行治療,如中止蛋白質攝入、靜脈補液、肌注大劑量維生素B12。
對所有甲基丙二酸尿症患者應首先進行大劑量維生素B12試驗治療,1mg/d肌肉注射,3~5d,對照治療前後尿甲基丙二酸濃度,判斷對維生素B12的反應性。通過大劑量維生素B12試驗治療不僅可以爭取治療時機,挽救維生素B12反應型患者,亦有助於病型診斷,指導長期治療。
維生素B12無效型以飲食治療為主,理想方式為限制天然蛋白質,補充去除異亮氨酸、纈氨酸、蛋氨酸、蘇氨酸的特殊治療奶粉,嬰幼兒期天然蛋白質每日攝入量應控制在1.0~1.2g/kg。維生素B12有效型其維生素B12長期維持劑量為1mg每周至每月肌肉注射1次或每日口服甲基鈷胺素500~1000μg,中等量蛋白質攝入,使血、尿甲基丙二酸濃度維持在理想範圍。
由於甲基丙二酸、丙酸等有機酸蓄積,生成相應酯醯化肉鹼,導致肉鹼消耗增加,補充肉鹼可促進酯醯肉鹼排泄,增加機體對自然蛋白的耐受性,不僅有助於急性期病情控制,亦可有效地改善預後。急性期可採用靜脈或肌內注射肉鹼每日100~200mg/kg,緩解期每日30~60mg/kg,長期維持。對於合併同型半胱氨酸血症的患者,應給予甜菜鹼(betaine)補充治療(1000~3000mg/d),降低血液同型半胱氨酸濃度,改善患兒神經系統情況。
預防後遺
該病的預後取決於病型、發現早晚及長期治療的合理性。但有人認為發病的早晚與中樞神經系統的預後及生命的長短無關。鈷胺素合成缺陷者發病較變位酶缺陷者晚,經過治療後大多能存活。mut0型預後最差,維生素B12不反應型預後不佳,Cb1A型預後最好。神經功能損害的輕重與高氨血症、代謝性酸中毒等持續時間的長短有關,通過早期診斷和治療可降低死亡率,減少神經系統後遺症。
病例舉證
 患兒
患兒例1
男,71H。因呼吸困難,拒乳11h入院。系第1胎,第1產,孕40+2周順產,新法接生,生後Apgar評分1min9分,5、10min均為10分。生後10h開奶,母乳餵養,吸吮有力。生後60h出現拒乳、呼吸困難,遂入院。父母非近親結婚,母孕期體健。體檢:體溫36.3℃,呼吸66次/min,脈搏140次/min,體質量2700g。足月新生兒貌,反應差,哭聲弱,全身皮膚中度黃染,前囟平坦,口周發紺,呼吸急促,三凹征陽性,雙肺呼吸音粗,未聞及乾、濕性囉音,心音有力,律齊,腹軟,肝肋下3cm,質軟,四肢肌張力低,新生兒反射減弱。頭顱CT示後縱裂內可見稍高密度影,考慮為蛛網膜下腔少量出血。血常規、血CRP、腦脊液常規均正常,尿蛋白(+),血清BUN水平為12.4mmol/L,Cr160.0mmol/L,肝功能異常,血清ALT35.1mmol/L,AST91.6mmol/L,血糖5.6mmol/L,血氨39.0μmol/L,乳酸3.3mmol/L,β羥丁酸1.62mmol/L,丙酮酸218μmol/L。血氣分析:pH7.084,二氧化碳分壓[p(CO2)]1.33kPa,氧分壓[p(O2)]14.27kPa,實際碳酸氫鹽(HCO-3)2.6mmol/L,剩餘鹼(BE)-27mmol/L,Na+150mmol/L,K+5.6mmol/L,Ca2+1.0mmol/L。予糾酸、支持治療,1d後複查血氣分析、電解質恢復正常。入院第2天抽搐3次,予水合氯醛止抽後未再抽搐。北京大學第一醫院氣相色譜分析提示尿液甲基丙二酸水平為249.32μg/(mg·Cr),32羥基丙酸水平為15.46μg/(mg·Cr)、甲基枸櫞酸水平為5.78μg/(mg·Cr);血漿同型半胱氨酸4.22μmol/L,尿液同型半胱氨酸水平為8.74μmol/L,支持診斷甲基丙二酸尿症。予限制天然蛋白質,補充特殊胺基酸粉,維生素B12肌內注射1mg/次,1次/周;葉酸5mg/d;左鏇肉鹼500mg/次,2次/d。治療1個月患兒一般情況好轉,飲食、睡眠好,呼吸平穩,反應好,哭聲響亮,心肺肝脾未見異常,四肢肌張力和反射正常。血氣分析和肝腎功能正常。出院後隨訪至今,患兒智力和運動發育明顯落後,3個月後複查頭顱CT示前縱裂、外側裂增寬,腦白質變薄。
例2
女,27h。因吃奶差24h入院。系第1胎,第1產,孕40+1周順產,新法接生,生後1、5和10minApgar評分均為10分,羊水Ⅲ度污染,量中等,胎盤和臍帶無異常。生後3h開奶,混合餵養,吃奶差,無嘔吐,反應欠佳,遂轉入兒科。父母非近親結婚,母孕期體健。體檢:體溫36.5℃,呼吸46次/min,脈搏140次/min,體質量2800g。足月新生兒貌,反應欠佳,全身皮膚無黃染,前囟平坦,心肺聽診正常,腹軟,肝肋下1cm,質軟,四肢肌張力正常。入院後吃奶好轉,20~30mL/次,2~3h/次,入院第2天,出現頻繁呼吸暫停,反應差,面色發紺,呼吸急促,三凹征陽性,四肢末端稍涼,四肢肌張力減低。急查血氣分析顯示:pH6.991,p(CO2)7.47kPa,p(O2)7.47kPa,HCO-39.2mmol/L,BE-24mmol/L,Na+136mmol/L,K+5.1mmol/L,Ca2+1.1mmol/L,血糖5.2mmol/L。頭顱CT示雙側額葉及雙枕葉腦白質密度彌散性減低,腦後縱裂密度稍增高,邊緣稍模糊。血WBC3.2×109L-1,RBC5.6×1012L-1,Hb177g/L,PLT10×109L-1。腦脊液常規和生化、血CRP、肝腎功能正常。予反覆糾酸,共予碳酸氫鈉60mL,及呼吸支持等治療。代謝性酸中毒呈進行性加重,考慮先天性代謝性疾病:胺基酸代謝異常。經積極搶救14h後死亡,1周后北京協和醫院氣相色譜2質譜分析報告為甲基丙二酸尿症。
世界罕見病名錄
| 罕見病,為患病人數占總人口的0.65%~1%的疾病,許多罕見病的病因在醫學上並不十分明確,所以罕見病的治療是醫學上的一大難題,本任務盤點一下一些罕見病。 |
