簡介
Down綜合徵又稱先天愚型或21三體綜合徵,屬常染色體畸變,是小兒染色體病中最常見的一種,活嬰中發生率約1/(600~800),母親年齡愈大,本病的發病率愈高。60%患兒在胎兒早期即夭折流產。
Down綜合徵很早就引起了一些人類遺傳學家的注意。
 Down綜合徵
Down綜合徵例如1932年Waardenburg曾認為“先天愚型”患者千篇一律的一整組症狀可能是一個具有特定染色體畸變,他建議檢查先天愚型患者是否有“染色體缺陷”或“不分離”,又或“染色體重複”,但由於當時還沒有合適的方法對他的這一結論加以驗證,所以對先天愚型的研究未能深入下去。直到建立了人類染色體分析技術後,1959年春,法國的細胞遺傳學家Lejeune等分析了9例先天愚型患兒經培養的成纖維細胞的染色體,首先證實此病為21三體。
發生率
新生兒的DS發生率約為1/1000~2/1000,據估計我國目前大約有60萬以上的DS患兒,按目前的出生率,我國平均20分鐘就有一例DS患兒出生,全國每年出生的DS患兒可多達27000例左右。發生率隨母親生育年齡的增高而增高,尤其當母親年齡大於35歲時,發生率明顯增高(圖1)。
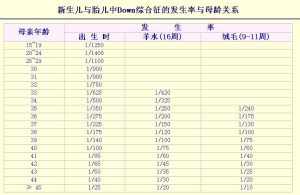 圖1——新生兒與胎兒中Down綜合徵的發生率與母齡關係
圖1——新生兒與胎兒中Down綜合徵的發生率與母齡關係這是由於產婦年齡越大,人體包括卵巢所承受的各種有害物質的影響也就越多,這些因素都會導致卵細胞異常,導致染色體在細胞分裂過程中出現不分離現象。有資料表明父親的年齡也與本病發病率有關,環境污染及接觸有害物質均可造成精子的老化和畸形,當父親年齡超過39歲時,出生患兒的風險將會增高。
表型特徵
DS患者有多種臨床表現,其主要表現為智力低下(患者的IQ值在20~60之間,平均為40~50歲)、發育遲緩和特殊面容。一般情況下,DS患者都具有一些明顯的、特殊的微小畸形特徵(圖2)。
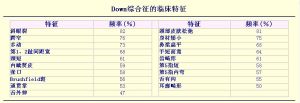 圖2——Down綜合徵的臨床特徵
圖2——Down綜合徵的臨床特徵儘管一些DS患者都因具有這些典型的特徵而易於被識別,但並不是所有的患者都表現出這些特徵,而具有圖2中所列的所有特徵的DS患者是非常罕見的。
Down綜合徵一般特點包括:
①這是一種很明確的綜合徵,儘管在症狀上有所不同,但並不會影響診斷;
②多數情況下,都是新發生的、散在的病例,家庭中很少有一個以上的該病患者;
③同卵雙生具有一致性,但偶爾也會有例外,這可能是由於在形成其中一個時,發生了染色體丟失;
④男性患者沒有生育力,而極少數女性患者可生育;
⑤隨母親年齡增加該病的發生率也升高,尤其當母親大於35歲時發病率明顯升高;
⑥病人的預期壽命短,且患者到中年時大腦呈現澱粉樣斑,與Alzheimer病相符,伴痴呆症狀;易感染表明免疫功能缺陷,先天性心臟病也增加,用抗生素和心臟外科手術治療可以延長病人的壽命;
⑦表型特徵的表現度不同;
⑧急性白血病死亡率增加了20倍,其原因尚不清楚。
遺傳分型
根據患者的核型組成不同,可將Down綜合徵分為三種遺傳學類型。
(一)游離型
游離型(21三體型)即標準型。據統計,此型約占全部患者的92.5%。核型為47,XX(XY),+21。此型的發生絕大部分與父母核型無關,它是生殖細胞形成過程中,在減數分裂時不分離的結果。染色體不分離發生在母方的病例約占95%,另5%見於父方,且主要為第一次減數分裂不分離。減數分裂不分離的機制還有待進一步研究,有研究表明可能與染色體支架蛋白-拓撲異構酶Ⅱ(topoⅡ)的活性改變有一定關係。此外,不能排除某些表型正常的母親實際是21三體細胞很少的嵌合體,如果其生殖細胞中嵌合21三體細胞,她們的子女就有可能遺傳獲得額外的21號染色體,特別是較年輕的、有過1個以上的21三體型孩子的母親。
(二)易位型
此型約占5%,增加的一條21號染色體並不獨立存在,而是與D組或G組的一條染色體發生羅伯遜易位,染色體總數為46,其中一條是易位染色體。最常見的是D/G易位,如核型為46,XX(XY),-14,+t(14q21q),其次為G/G易位,如核型為46,XX(XY),-21,+t(21q21q)。患者的易位染色體,如果是由親代傳遞而來的,其雙親之一通常是表型正常的染色體平衡易位攜帶者(balancedtranslocationcarrier),其核型為45,-21,+t(Dq21q)或45,+t(Gq21q)。染色體平衡易位攜帶者在生殖細胞形成時,理論上經減數分裂可以產生6種類型的配子(圖3),
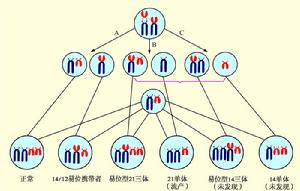 圖3——14/21染色體平衡易位攜帶者及其子女核型圖解
圖3——14/21染色體平衡易位攜帶者及其子女核型圖解但實際上只有4種配子形成,故與正常個體婚配後,將產生4種核型的個體。由此可見,染色體平衡易位攜帶者雖外表正常,但其常有自然流產或死胎史,所生子女中,約1/3正常,1/3為易位型先天愚型患兒,1/3為平衡易位攜帶者。但如果父母之一是21/21平衡易位攜帶者時,1/2胎兒將因核型為21單體而流產,1/2核型為46,-21,+t(21q21q),因此活嬰將100%為21/21易位型先天愚型患兒。所以21/21平衡易位攜帶者不應生育。
(三)嵌合型
此型較少見,約占2%。此型產生的原因
一是由於生殖細胞減數分裂不分離,繼而因分裂後期染色體行動遲緩引起部分細胞超數的染色體發生丟失而形成含有47,+21/46兩個細胞系的嵌合體,由此形成的嵌合體的發生率與標準的三體型一樣,隨母親年齡的增加而增加。
二是合子後(post-zygotic)有絲分裂不分離的結果。
如果第一次卵裂時發生不分離,就會產生47,+21和45,-21兩個細胞系,而後一種細胞很難存活,因此,導致嵌合體的不分離多半發生在以後的某次有絲分裂,所有嵌合體內都有正常的細胞系。不分離發生得越晚,正常細胞系所占比例就越多,則此患者症狀就越輕。有研究表明,有絲分裂過程中不分離形成的嵌合體與母親年齡無關,在已有的報導中由於有絲分裂不分離形成的嵌合體估計占17%~30%。因本型患者的體細胞中含有正常細胞系,故臨床症狀多數不如21三體型嚴重、典型。如47,+21細胞系比例低於9%時,一般不表現出臨床症狀。
分子機制
(一)21號染色體的分子解剖學
2000年5月,由日、德等國科學家通力合作的人類21號染色體DNA序列測定工作完成,論文在Nature發表。21號染色體是人類染色體中最小的一條,由51×106bp組成,約長46cM,包含600~1000個基因,占整個人類基因組的1.7%。用染色體顯帶技術顯示21號染色體短臂分1區3帶,長臂分2區,1區僅有1帶,2區分2帶,各帶又可分出亞帶,2區2帶可分為3個亞帶。
用細胞遺傳學和分子生物學方法對多例DS病人進行分析,結果表明有一短的重複序列導致部分DS的表型。現已證明這是大約為400kb的DNA。
(二)21號染色體上與DS表型相關的基因
通過對部分21三體的基因型與表型關係的研究,現已將DS的24種特徵定位在21號染色體的6個小區域,其中2個區域尤為引人關注
①D21S55:表達13種特徵的最小區域。13種特徵分別是:智力障礙、身材矮小、肌張力下降、關節鬆弛和9種面貌特徵:鼻樑扁平、舌外伸、齶弓高、窄齶、耳廓畸形、手掌寬且短、第五指短且彎、足第一、二趾間距寬;
②D21S55-MX1:表達6種外貌特徵(眼裂斜、內眥贅皮、Brushfield斑-虹膜周圍小白斑、通貫手、指紋尺箕和小魚際肌無側環)的最小區域(表13.3);D21S55在DS的發病機制中起重要作用,在21q22.2跨0.4-3kb。D21S55及21q22.3遠端被稱為DS關鍵區(Down syndrome critical region,DCR)。
一些研究已顯示與DS發病有關的基因可能是一些結構基因或調控基因,但具體作用機制尚不太清楚。
1.與智力發育遲緩相關的基因
(1)DS細胞黏附分子(Down syndrome cell adhesion molecule,DSCAM)基因定位於21q22.2-22.3,長75Mb,編碼一種細胞黏附分子。該基因不同程度地表達在成人腦組織中,小鼠DSCAM基因的組織原位雜交顯示該基因在中樞及外周神經中都有表達,提示DSCAM參與神經系統分化,並與DS中樞和外周神經缺陷有關。RT-PCR顯示該基因也表達在人胚胎和7.5~10周胎兒心臟組織中,其過度表達與DS的先天性心臟病發生也有關係。
(2)活性依賴性神經保護蛋白(Activity dependent neuroprotive protein,ADNP)基因多在海馬、大腦皮質和小腦中表達,ADNP由828個胺基酸構成,PI為5.99,廣泛分布於所有器官。經多項研究顯示ADNP是一新型的熱休克蛋白,它為DS提供一個該機體所缺乏的保護作用。
(3)DSCR1基因位於21q21.1-22.2,在胎兒及成人心臟中高度表達。一系列實驗顯示DSCR1在體內參與調節calcineurin的活性,而calcineurin又可調節許多生理過程,如神經遞質和激素釋放、突觸形成和基因轉錄等,因此推測DSCR1可能與DS的學習和行為變化有關。
2.與先天性心臟缺陷(congenital heart defects,CHD)有關的基因
(1)COL6A1/2基因位於21q22.3,該區編碼的蛋白包含一個von Willebrand 因子的基因,該基因具有連線膠原的特性,可與膠原Ⅵ四聚體連線構成特徵性串珠狀絲。DS編碼的該蛋白若有胺基酸改變,發生先天性心臟缺陷的機率會增加。
(2)KCNE-2基因位於21號染色體長臂,與KCNE-1基因相似。它在成人心臟中高度表達,在骨骼肌中也有少量表達,轉錄長度為35kb,編碼123個胺基酸殘基的開放閱讀框架,在胺基酸水平,KCNE-2與人類Isk蛋白高度相似。Isk是一膜蛋白,當與KCNQ-1或KCNE-1形成複合物後,即構成被緩慢激活的電壓依賴型K+通道。KCNE-2位於細胞膜上,N-端位於細胞外,因與Isk相似程度高及亞細胞水平表達的組織特異性,可認為KCNE-2參與形成心臟電壓依賴型K+通道,其異常可能與CDH有關。
3.與白血病有關的基因
白血病在DS患者中的發生率較正常人高約20倍,最常見的類型是急性淋巴細胞白血病(ALL)。FISH顯示DS病人的21號染色體上的21q11.2和21q22兩個區域有潛在的結構改變。在6/8的有明顯白血病症狀的DS患者中發現D21S65-D21S55和D21S19-D21S219/D21S220間位點缺失或部分缺失。21q22上的AML1基因易位也常產生白血病。AML基因又稱CBFα或PEBP2α基因,是異二聚體轉錄因子基因家族成員之一,在人類已鑑定出3個AML基因:AML1位於21q22.1,AML2位於1p36,AML3位於6p21。AML1在造血中起關鍵作用,其表達有組織特異性,通過可變剪下產生編碼區大小不同的一組mRNA(188~480個胺基酸殘基),其中一些蛋白質在體內可以抑制腫瘤生長和來自畸胎瘤ES細胞分化。
4.與肌張力低下有關的基因
肌張力低下幾乎出現於所有DS患者中,其發生主要與位於21號染色體上DSCR區D21S335和D21S337之間MNBH/DYRK1基因有關。MNBH基因由17個外顯子組成,跨越150kb,通過不同啟動方式產生兩種轉錄單位MNBHa和MNBHb,MNBHa在各組織中廣泛表達,而MNBHb只表達於心臟和骨骼肌中。雖然兩種啟動方式在成人心臟和骨骼肌中都存在,但由TATA式啟動子開始的表達只被控制在肌肉組織中。MNBHb在肌肉組織中表達模式提示MNBH與影響大多數DS患者的肌張力低下的病理生理有關。
DS還伴有其他疾病,如:內分泌異常、腸道異常和免疫缺陷、耳聾等先天性缺陷,其基因型與表型的關係還尚待研究。
21號染色體DNA測序的完成,無疑將加快21號染色體基因功能的研究,對揭示DS及其他疾病的分子病因,更快、更精確地診斷DS,並且在分子病理學上進行干預性治療,都具有深刻的意義。
診斷
(一).臨床篩查
DS臨床診斷的正確率甚高,90%以上的病例根據典型的DS面容及智力低下即可作出診斷,如DS在新生兒期除特殊面容外還有肌張力低、第三囟門、通貫手、小指短而內彎、小指一條褶紋、足跖溝、足第一二趾間距寬(草鞋足)等易被觀察的臨床指征。但新生兒期患者有時面容不夠典型,又難以觀察智力反應,故易被忽視而漏診,所以還應進行染色體分析予以確診,這將對遺傳諮詢提供依據,特別是查出易位型患者,追查其家系染色體,檢出平衡易位攜帶者,可預防患兒的再出生。
(二).染色體檢查
絕大部分為21三體型,少數為嵌合型和易位型。染色體檢查對本病的診斷是決定性的。
(三).血液學改變
DS病人白細胞計數正常,中性粒細胞相對增多,分葉少且呈核左移。新生兒在感染時易出現類白血病反應,血紅蛋白F和血紅蛋白A2升高,無需治療,能自發恢復,但常在1~2年後出現真正的白血病。
(四).酶的改變
過氧化物歧化酶(SOD-1)基因定位於21q22。21三體綜合徵患者細胞中SOD-1的含量較正常人高50%。中性粒細胞的鹼性磷酸酶活性也較正常人高50%,其基因也定位於21號染色體上。
治療
目前對促進智慧型發育無特效藥物,可試用γ-氨酪酸、谷氨酸、維生素B6、葉酸等,對促進小兒精神活動、提高智商可能有一些作用。對先天性心臟病,可用抗生素和心臟外科手術治療以延長病人的壽命。
預防
(一).為防止DS患兒的出生,對35歲以上的孕婦、30歲以下但生育過DS患兒的孕婦或其雙親之一是平衡易位攜帶者或嵌合體者應作產前檢查,如取孕16~20周的羊水細胞或9~12周的絨毛膜細胞作染色體檢查,如胎兒為21三體,則應終止妊娠。
(二).年齡在30歲以下,且生過21三體患兒及一級親屬中有DS患者或有平衡易位攜帶者的婦女,應作染色體檢查。如孕婦為平衡易位攜帶者應作產前檢查,21/21易位攜帶者則不應生育。
(三).此外育齡婦女妊娠前後應避免接受較大劑量射線照射,不隨便服用化學藥物,預防病毒感染。
預後
3/4的DS胎兒在懷孕期已自發流產,且大部分發生在妊娠3個月內,僅約1/4胎兒能活到出生。患者智力低下,缺乏抽象思維能力,精神運動性發育缺陷,但許多患者經過訓練可以學會讀和寫,以及一些基本的生活技能,如穿衣、吃飯等。一些人還可以達到接近邊緣的社會適應力。但絕大部分人都不能靠自己在社會上活動。DS患者在30多歲時智慧型便開始下降,通常伴隨著社交能力的逐漸喪失和情緒衰退,這些表現是Alzheimer病的症狀,但這些症狀出現過早。隨著醫療水平的不斷提高,現在的DS患者的生存期比以前感染未能被控制時要長。許多人可以活到成年。但一般壽命比正常人短,只有8%的患者活過40歲。
遺傳諮詢
DS中極大部分為典型的三體型,發生率隨母親生育年齡的增高而增高,因此高齡孕婦(大於35歲)的胎兒應作產前診斷。在美國、加拿大等已開發國家多年來對35歲以上的孕婦普遍都作產前診斷,以預防DS患兒的出生。但由於絕大部分孕婦在35歲以下,而事實上未經產前診斷的35歲以下的孕婦所生的DS患兒占了80%,然而又不可能對全部孕婦作羊水穿刺檢查。所以近些年來,國外提出於孕中期用孕婦血清標記物篩查DS胎兒。由於DS胎兒的孕婦血清的AFP(甲胎蛋白)及UE3(雌三醇)低於平均水平,HCG(絨毛膜促性腺激素)高於平均水平,因此對妊娠期(孕15~21周)的孕婦測定此三項值,即所謂的“三聯篩查”,再結合孕婦年齡,計算出危險度,以決定是否行產前診斷,其檢出率為48%~83%,假陽性率約5%。1995年Wallance等發現在孕早期(孕11~13周)DS胎兒的母血清中二聚體抑制素A(由黃體與胎盤分泌的一種異二聚糖蛋白)含量明顯升高,也可篩查DS胎兒,是一種敏感、特異的新方法,且可提早診斷,減輕孕婦痛苦,較三聯篩查更具優越性;其檢出率為65%,假陽性率為4%。
對於各種平衡易位型攜帶者,其遺傳後果也不完全相同。Dq21q平衡易位的攜帶者理論上通過減數分裂可以形成6種配子,但受精後除不能發育者外,僅可產生三種胎兒:正常胎兒、平衡易位者、易位型三體患兒,即產生患兒的風險為33.3%。但實際風險較低,其再發風險可根據經驗估計,這與雙親哪一方為攜帶者有關。Dq21q易位攜帶者若是母親,生育患兒的風險為10%~15%;如為父親,則風險為5%或更小。21q22q易位的情況與之大體相同,但易位染色體由父方傳遞的百分比比D/G易位多,風險率在10%以下。21q21q易位攜帶者雖不常見,但尤為重要,因為其只能產生三體或單體的合子,即不可能有正常表型的胎兒;因單體不能存活,故此種易位型攜帶者的後代將100%為三體型患兒,所以21q21q攜帶者不宜生育。從上述幾種易位型攜帶者子代再發風險率看,均明顯高於典型的三體型,尤其是21q21q攜帶者,因此,檢出平衡易位攜帶者的雙親具有重要意義。
