配位化學
正文
研究金屬的原子或離子與無機、有機的離子或分子相互反應形成配位化合物(簡稱配合物)的特點以及它們的成鍵、結構、反應、分類和製備的學科。例如,【Fe(CO)5】、【Cu(NH3)4】2+和【Ni(CN)4】2-這三種配合物,分別由起中心作用的原子Fe或離子 Cu2+和Ni2+(統稱中心原子)與圍繞它們的稱為配體的分子CO、NH3或離子CN-所組成。配合物在溶液中會發生離解,但仍保持其本體。配合物的電荷可以是正、零或負,由中心原子和配體所帶電荷決定。在中心原子周圍的配體總數稱為配位數。簡史 最早有記載的配合物是18世紀初用作顏料的普魯士藍, 化學式為 K【FeⅡ(CN)6FeⅢ】 。1798年發現CoCl3·6NH3。CoCl3和NH3都是穩定的化合物,在它們結合成新的化合物後,其性質與組分化合物不同。這一發現開創了配位化學的研究。
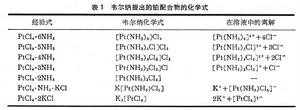
19世紀就發現了更多的鈷氨配合物和其他配合物。1893年瑞士化學家A.韋爾納首先提出這類化合物的正確化學式及配位理論。他在配合物中引進副價的概念,提出元素在主價以外還有副價。例如,在一系列鉑(Ⅳ)的配合物中鉑的主價為+4,副價為+6,由此可解釋這些鉑配合物的存在和離解(表 1)。生成的離子數目由溶液電導和游離氯離子的分析確定。由於韋爾納的出色工作,他於1913年獲得諾貝爾化學獎。
 配位化學
配位化學價鍵理論 主要是由L.C.鮑林發展起來的。該理論認為配合物是在路易斯鹼(配體)和路易斯酸(金屬或金屬離子)之間反應生成(見酸鹼理論),在配體和金屬之間有配位鍵生成(不必全是配位鍵)。配體上的電子對轉到金屬的雜化原子軌道上。
晶體場理論 認為金屬-配體鍵是由點電荷之間的反應生成,把配體看作點電荷或偶極子,因而影響金屬離子的部分已占的d軌道能量,並用來說明其成鍵結構。
分子軌道理論 認為電子圍繞整個配合物體系的分子軌道運動,它綜合了價鍵和晶體場理論,是當前用得最廣泛的理論。
配合物的穩定性 與金屬離子和配體有關。由於配合物的生成主要是在荷正電的金屬離子和配體陰離子或偶極分子之間進行的,金屬離子的離子勢(陽離子電荷與其半徑之比)愈大,相同配體的配合物愈穩定。配合物的穩定性還與配體陰離子的可極化性有關。在一定限度內,陰離子的可極化性愈大,配體也愈易成為電子給體。例如,對於第四周期從Mn2+到Zn2+的二價金屬離子,其配合物穩定性按F<O<N>S>P次序變化。對於d8或d10結構的貴金屬,其配合物的穩定性,按P>S》N>O>F《Cl<Br<I的次序變化。
影響配合物穩定性的還有螯合作用,即雙齒以上的配體在多於一個的位置上與金屬離子連線成環。通常,螯合程度增加時,配合物的穩定性也就增加,例如乙二胺配合物的穩定性要比氨配合物大。
配合物的類型 按中心原子分類有單核配合物和多核配合物;按配體分類,常見的有水合配合物、鹵合配合物、氨配合物、氰配合物、金屬羰基合物等;按成鍵類型分類有經典配合物(σ 配鍵)、簇狀配合物(金屬-金屬鍵)、烯烴等不飽和配體的配合物(π-σ鍵和π-π*反饋鍵)、鐵茂等夾心、穴狀、籠狀配合物(離域共軛配鍵)等;按學科類型分類有無機配合物、有機金屬化合物、生物無機化合物等。各種分類之間又有交叉。
水合配合物 簡稱水合物。為金屬離子與水分子形成的配合物。幾乎所有金屬離子在水溶液中均可形成水合配合物,例如【Cu(H2O)4】2+和【Cr(H2O)6】3+,常見的配位數是4和6。當其他配體加到金屬離子水溶液中時,發生對水的置換作用,生成其他配合物。在這些置換反應中,金屬的配位數一般保持不變。但並不總是如此,例如,將CN-離子加到含有【Ni(H2O)6】2+的水溶液中,最後生成【Ni(CN)4】2-和【Ni(CN)5】3-;將Cl-離子加到【Fe(H2O)6】3+中可產生【FeCl4】-等。
鹵合配合物 金屬離子與鹵素(氟、氯、溴、碘)離子形成的配合物。絕大多數金屬離子均可生成鹵合配合物,例如K2【PtCl4】和Na3【AlF6】。鹵合配合物的穩定性變化範圍很大,可按軟硬酸鹼理論的概念把金屬離子和配體分類。金屬離子分成硬酸和軟酸。硬酸是指那些體積小、電荷高和沒有容易變形或移動的價殼層電子的金屬離子,例如Be2+、Mg2+、Al3+、Sc3+、La3+、Cr3+、Co3+和Fe3+等;軟酸是指那些體積大、電荷低和具有容易變形或移動的價殼層電子的金屬離子,例如Cu+、Ag+、Au+、Cd2+、Hg2+、Pd2+和Pt2+等。配體也可分為不易被極化的硬鹼和易被極化的軟鹼,鹵素中的F-和Cl-為硬鹼,I-為軟鹼,Br-則介於二者之間。硬酸容易與硬鹼結合,軟酸容易與軟鹼結合。
硬酸類金屬的鹵合配合物在水溶液中的穩定性是按I<Br<Cl《F的次序遞增,軟酸類金屬鹵合配合物的穩定性則按F《Cl<Br<I的次序遞增。
金屬羰基合物 為金屬與一氧化碳結合的產物。在金屬羰基合物中金屬的氧化態都很低,有的羰基合物中金屬氧化態為零,如Ni(CO)4;有的呈負氧化態的,如Na【Co(CO)4】;有的呈正氧化態,如【Mn(CO)5Br】。這些都是單核羰基合物。還有多核金屬羰基合物,如Fe2(CO)9和Fe3(CO)12(見羰基金屬)。
簇狀配合物 即簇狀化合物。含有至少兩個金屬,並含金屬-金屬鍵的配合物,例如Fe3(CO)12、Co4(C5H5)4H4、(W6Cl12)Cl6等。能夠生成簇狀化合物的金屬主要是過渡金屬(見過渡元素),它們的生成趨勢與該金屬在周期表中的位置、氧化態以及配體性質等條件有關。一般地說,第一過渡系的元素形成簇狀配合物的能力比相應的第二、第三過渡系元素差。在同種元素中,低氧化態的容易形成簇狀配合物(見原子簇金屬化合物)。
有機金屬化合物 或稱金屬有機化合物,為有機基團與金屬之間生成碳-金屬鍵的化合物。許多有機金屬化合物往往以配合物的形式存在,這種配合物有兩種類型:①金屬與碳直接鍵合的σ 鍵(見共價鍵)有機金屬化合物,包括烷基金屬【如(CH3)6Al2】、芳基金屬(如C6H5HgCl)、乙炔基金屬(如HC呏C─Ag)等類化合物。大多數這類化合物,除有機配體外還可含有其他配體,例如CO、CN-、PR3(R為烷基)等;②π鍵有機金屬化合物,包括烯烴、炔烴、芳烴、環戊二烯基等配合物,例如蔡斯鹽K【PtCl2(C2H4)】,其結構如
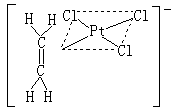 。在Pt和CH2=CH2之間是π-σ及π-π*鍵合。後又陸續製得銀、銅、鈀、釕等其他金屬離子的烯烴配合物。又如二茂鐵(C5H5)2Fe(見結構式a
。在Pt和CH2=CH2之間是π-σ及π-π*鍵合。後又陸續製得銀、銅、鈀、釕等其他金屬離子的烯烴配合物。又如二茂鐵(C5H5)2Fe(見結構式a ),金屬原子被夾在兩個平行的碳環體系之間,稱為夾心化合物。除了環戊二烯基外,還有其他不飽和環狀配體的夾心化合物,如二苯鉻(C6H5)2Cr(b)。生成夾心化合物的元素,主要是過渡元素中的第Ⅳ到第Ⅶ副族元素和除鉑以外的第Ⅷ族元素,以及鑭系元素和錒系元素(見金屬有機物的鍵型)。
),金屬原子被夾在兩個平行的碳環體系之間,稱為夾心化合物。除了環戊二烯基外,還有其他不飽和環狀配體的夾心化合物,如二苯鉻(C6H5)2Cr(b)。生成夾心化合物的元素,主要是過渡元素中的第Ⅳ到第Ⅶ副族元素和除鉑以外的第Ⅷ族元素,以及鑭系元素和錒系元素(見金屬有機物的鍵型)。 生物無機化合物 生物配體與金屬的配合物。它們在生物體中含量雖然不多,但在一系列有機體的生命活動中具有很重要的作用。多種金屬蛋白質是金屬酶,常能催化某種反應,例如含鋅的碳酸酐酶可催化二氧化碳水合反應或其逆反應;含鐵蛋白質(肌紅蛋白、血紅蛋白和蚯蚓血紅蛋白)、含銅蛋白質(血青肌)和含釩蛋白質(血釩蛋白)能貯存和輸送氧。其他重要的生物無機配合物還有葉綠素、維生素B12等(見生物無機化學)。
配合物的製法 許多配合物可由其組成化合物直接加成製得,例如由氣相的BF3和NH3反應製備【F3B·NH3】。
由一種配體取代另一種配體,也是常用的一種製備配合物的方法。例如用乙二胺置換【Co(NO2)6】3-中的硝基,得到順式-【Co(en)2(NO2)2】+(en為乙二胺)。
當金屬離子具有不同氧化態時,可用氧化還原反應製備不同價態金屬的配合物。高氧化態金屬離子配合物,有時可在氧化劑存在下,由配體與低氧化態金屬離子作用製得。例如在空氣或氧存在下,氨與鈷(Ⅱ)鹽水溶液作用,生成鈷(Ⅲ)氨配合物。低氧化態金屬離子配合物,有時可在還原劑存在下,由配體與高氧化態金屬鹽作用製得。例如由CoCO3和H2及CO反應製備Co2(CO)8,H2是還原劑,CO是配體兼還原劑。
利用熱分解方法,通過有控制地加熱一些配合物可製得另一些配合物,例如加熱 【Cr(en)3】Cl3可以製得順式-【Cr(en)2Cl2】Cl。
配合物主要反應 酸鹼反應 由於水合金屬離子離解,生成質子,金屬離子在水溶液中通常顯酸性,例如:
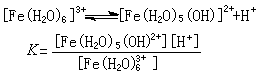
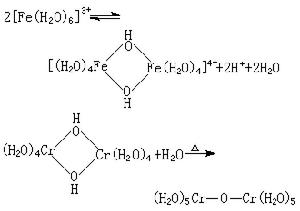
取代反應 指配體取代配合物中另一種配體的反應。根據取代反應的快慢,常把配合物分為活性配合物和惰性配合物。金屬水合配合物中水被取代的反應速率常作為活性或惰性的衡量標準。取代基可以是水分子或其他配體,如為前者,可用標記原子18O(以符號*O表示)示蹤。例如:
【Al(H2O)6】3++6H2*O匑【Al(H2*O)6】3++6H2O
各種水合金屬離子的配位水分子與溶液本體中水分子取代速率相差很大。例如鹼金屬水合離子的取代反應速率常數為108~1010秒-1,而鋁和鎵的水合離子則為1~103秒-1。離子的大小和所帶電荷的多少對反應速率有明顯的影響。電荷和結構相同的離子,半徑愈大,交換得愈快;離子大小相同者,電荷愈高,交換得愈慢。其他配體取代水合金屬離子中的配位水分子的反應速率很少取決於配體的性質,而常與水分子的交換速率一致,即取決於水合金屬離子的性質。電子轉移反應 指兩配合物之間發生電子轉移的反應。例如,將 【*Fe(CN)6】4-(*Fe為標記原子)溶液與【Fe(CN)6】3-混合,則前者失去一個電子,後者得到一個電子,其反應為:
【*Fe(CN)6】4-+【Fe(CN)6】3-─→
【*Fe(CN)6】3-+【Fe(CN)6】4-
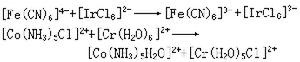
金屬的提取和分離 一些重要的濕法冶金過程要利用金屬配合物的形成,例如鎳、銅和鈷可用氨水溶液萃取。在核反應中產生的鈹,可用噻吩甲醯三氟丙酮的苯溶液萃取。氰化鈉的水溶液通常用於從礦石中分離金。一氧化碳可用於鎳的純化。
化學分析 配位反應在元素的重量分析、容量分析和光度分析中有廣泛的套用,主要用作顯色劑、指示劑、沉澱劑、滴定劑、萃取劑、掩蔽劑等。例如,以氟離子作為掩蔽劑,可與鐵(Ⅲ)生成無色而穩定的【FeF6】3-,在用碘量法測銅時避免了鐵(Ⅲ)離子的干擾;以二乙醯二肟作為沉澱劑,可使鎳和鈀同時生成螯合物沉澱,鎳的沉澱溶於酸,鈀的沉澱不溶,即可分離、鑑定鎳和鈀;以硫氰酸鹽作為顯色劑,可與Fe3+離子形成血紅色的配合物,即可鑑別Fe3+的存在。EDTA(乙二胺四乙酸)能與大多數的金屬離子生成穩定性不一的配合物,是滴定分析中的一種優良的滴定劑,通過控制溶液的pH值和加入掩蔽劑、解蔽劑,用EDTA可從各種金屬離子的混合溶液中分別定量地滴定出它們的含量,省去分離干擾元素的步驟。
催化作用 過渡金屬化合物能與烯烴、炔烴和一氧化碳等各種不飽和分子配位形成配合物,使這些分子活化,生成新的化合物。例如烯烴的氫甲醛化反應中,烯烴與氫和一氧化碳按照與鈷催化劑形成配合物的機理,最終生成醛(R為烷基):
RCH=CH2+CO+H2─→RCH2CH2CHO
有些金屬催化劑可把烯烴轉變為多聚體。例如,將氯化鈦(Ⅲ)和烷基鋁配位後,作為催化劑,可使烯烴定向聚合成高分子化合物。參考書目
戴安邦主編:《配位化學》(無機化學叢書),科學出版社,北京,1987。
J.C.Bailar,Jr.,ed.,The Chemistry of the Coor-dination Compounds, Van Nostrand Reinhold, New York,1956.
F.Basolo and R.C.Johnson, Coordination Chemis-try, The Study of Metal Complexes,Benjamin, New York,1964.
