
指標意義
機體對氧的攝取和利用是一個複雜的生物學過程。一般來講,判斷組織獲得和利用氧的狀態要檢測二個方面因素:組織的供氧量、組織的耗氧量。測定血氧參數對了解機體氧的獲得和消耗是必要的:
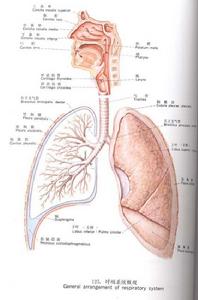
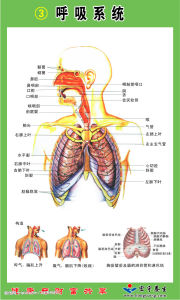 呼吸系統
呼吸系統1.氧分壓(partialpressureofoxygen,PO2)
為物理溶解於血液的氧所產生的張力。動脈血氧分壓(PaO2)約為13.3kPa(100mmHg),靜脈血氧分壓(PvO2)約為5.32kPa(40mmHg),PaO2高低主要取決於吸入氣體的氧分壓和外呼吸功能,同時,也是氧向組織彌散的動力因素;而PvO2則反映內呼吸功能的狀態。
2.氧容量(oxygenbindingcapacity,CO2max)
CO2max指PaO2為19.95kPa(150mmHg)、PaCO2為5.32kPa(40mmHg)和38℃條件下,100ml血液中血紅蛋白(Hb)所能結合的最大氧量。CO2max高低取決於Hb質和量的影響,反映血液攜氧的能力。正常血氧容量約為8.92mmol/L(20ml%)。
3.氧含量(oxygencontent,CO2)
CO2是指100ml血液的實際帶氧量,包括血漿中物理溶解的氧和與Hb化學結合的氧。當PO2為13.3kPa(100mmHg)時,100ml血漿中呈物理溶解狀態的氧約為0.3ml,化學結合氧約為19ml。正常動脈血氧含量(CaO2)約為8.47mmol/L(19.3ml/dl);靜脈血氧含量(CvO2)為5.35-6.24mmol/L(12ml%-14ml/dl)。氧含量取決於氧分壓和Hb的質及量。
4.氧飽和度(oxygensaturation,SO2)
SO2是指Hb結合氧的百分數。SO2=(氧含量–物理溶解的氧量)/氧容量×100%此值主要受PO2的影響,兩者之間呈氧合Hb解離曲線的關係。正常動脈血氧飽和度為93%-98%;靜脈血氧飽和度為70%-75%。
5.動–靜脈氧差(A-VdO2)
A-VdO2為CaO2減去CvO2的差值,差值的變化主要反映組織從單位容積血液內攝取氧的多少和組織對氧利用的能力。正常動脈血與混合靜脈血的氧差為2.68-3.57mmol/L(6ml%-8ml%)。當血液流經組織的速度明顯減慢時,組織從血液攝取的氧可增多,回流的靜脈血中氧含量減少,A-VdO2增大;反之組織利用氧的能力明顯降低、Hb與氧的親和力異常增強等回流的靜脈血中氧含量增高,A-VdO2減小。Hb含量減少也可以引起A-VdO2減小。
6.P50
P50指在一定體溫和血液pH條件下,Hb氧飽度為50%時的氧分壓。P50代表Hb與O2的親和力,正常值為3.47-3.6kPa(26-27mmHg)。氧離曲線右移時P50增大,氧離曲線左移時P50減小,比如紅細胞內2,3-DPG濃度增高1mmol/gHb時,P50將升高約0.1kPa。
低張性缺氧
低張性缺氧(hypotonichypoxia)指由PaO2明顯降低並導致組織供氧不足。當PaO2低於8kPa(60mmHg)時,可直接導致CaO2和SaO2明顯降低,因此低張性缺氧也可以稱為低張性低氧血症(hypotonichypoxemia)。
 缺氧CT圖
缺氧CT圖1、原因
低張性缺氧的常見原因為吸入氣體氧分壓過低、肺功能障礙和靜脈血摻雜入動脈血增多。
(1)吸入氣體氧分壓過低:因吸入過低氧分壓氣體所引起的缺氧,又稱為大氣性缺氧(atmospherichypoxia)。
(2)外呼吸功能障礙:由肺通氣或換氣功能障礙所致,稱為呼吸性缺氧(respiratoryhypoxia)。常見於各種呼吸系統疾病、呼吸中樞抑制或呼吸肌麻痹等。
(3)靜脈血分流入動脈:多見於先天性心臟病。
2、血氧變化的特點
①由於彌散入動脈血中的氧壓力過低使PaO2降低,過低的PaO2可直接導致CaO2和SaO2降低;
②如果Hb無質和量的異常變化,CO2max正常;
③由於PaO2降低時,紅細胞內2,3-DPG增多,故血SaO2降低;
④低張性缺氧時,PaO2和血SaO2降低使CaO2降低;
⑤動-靜脈氧差減小或變化不大。通常100ml血液流經組織時約有5ml氧被利用,即A-VdO2約為2.23mmol/L(5ml/dl)。氧從血液向組織彌散的動力是二者之間的氧分壓差,當低張性缺氧時,PaO2明顯降低和CaO2明顯減少,使氧的彌散速度減慢,同量血液彌散給組織的氧量減少,最終導致A-VdO2減小和組織缺氧。如果是慢性缺氧,組織利用氧的能力代償增加時,A-VdO2變化也可不明顯。
3、皮膚黏膜顏色的變化
正常毛細血管中脫氧Hb平均濃度為26g/L(2.6g/dl)。低張性缺氧時,動脈血與靜脈血的氧合Hb濃度均降低,毛細血管中氧合Hb必然減少,脫氧Hb濃度則增加。當毛細血管中脫氧Hb平均濃度增加至50g/L(5g/dl)以上(SaO2≤80%~85%)可使皮膚黏膜出現青紫色,稱為紫紺(cyanosis)。在慢性低張性缺氧很容易出現紫紺。紫紺是缺氧的表現,但缺氧的病人不一定都有紫紺,例如貧血引起的血液性缺氧可無紫紺。同樣,有紫紺的病人也可無缺氧,如真性紅細胞增多症患者,由於Hb異常增多,使毛細血管內脫氧Hb含量很容易超過50g/L,故易出現紫紺而無缺氧症狀。
血液性缺氧
血液性缺氧(hemichypoxia)指Hb量或質的改變,使CaO2減少或同時伴有氧合Hb結合的氧不易釋出所引起的組織缺氧。由於Hb數量減少引起的血液性缺氧,因其PaO2正常而CaO2減低,又稱等張性缺氧(isotonichypoxemia)。
1、原因
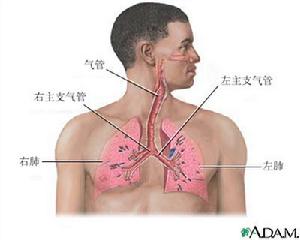 呼吸系統
呼吸系統(1)貧血:又稱為貧血性缺氧(anemichypoxia)。
(2)一氧化碳(CO)中毒:Hb與CO結合可生成碳氧Hb(carboxyhemoglobin,HbCO)。CO與Hb結合的速度雖僅為O2與Hb結合速率的1/10,但HbCO的解離速度卻只有HbO2解離速度的1/2100,因此,CO與Hb的親和力比O2與Hb的親和力大210倍。當吸入氣體中含有0.1%CO時,血液中的Hb可有50%轉為HbCO,從而使大量Hb失去攜氧功能;CO還能抑制紅細胞內糖酵解,使2,3-DPG生成減少,氧解離曲線左移,HbO2不易釋放出結合的氧;HbCO中結合的O2也很難釋放出來。由於HbCO失去攜帶O2和妨礙O2的解離,從而造成組織嚴重缺氧。在正常人血中大約有0.4%HbCO。當空氣中含有0.5%CO時,血中HbCO僅在20~30min就可高達70%。CO中毒時,代謝旺盛、需氧量高以及血管吻合支較少的器官更易受到損害。
(3)高鐵血紅蛋白血症:當亞硝酸鹽、過氯酸鹽、磺胺等中毒時,可以使血液中大量(20%~50%)Hb轉變為高鐵血紅蛋白(methemoglobin,HbFe3+OH)。高鐵Hb形成是由於Hb中二價鐵在氧化劑的作用下氧化成三價鐵,故又稱為變性Hb或羥化Hb。高鐵Hb中的Fe3+因與羥基牢固結合而喪失攜帶氧能力;另外,當Hb分子中有部分Fe2+氧化為Fe3+,剩餘吡咯環上的Fe2+與O2的親和力增高,氧離曲線左移,高鐵Hb不易釋放出所結合的氧,加重組織缺氧。患者可因缺氧,出現頭痛、衰弱、昏迷、呼吸困難和心動過速等症狀。臨床上常見的是食用大量新醃鹹菜或腐敗的蔬菜,由於它們含有大量硝酸鹽,經胃腸道細菌作用將硝酸鹽還原成亞硝酸鹽並經腸道黏膜吸收後,引起高鐵Hb血症,患者皮膚、黏膜(如口唇)呈現青灰色,也稱為腸源性紫紺(enterogenouscyanosis)。
在生理狀態下,血液中也有少量的高鐵Hb不斷形成,但可以通過體內還原劑如NADH、維生素C、還原型谷胱甘肽等還原為Fe2+,使正常血液中高鐵Hb含量限於Hb總量的1%~2%。高鐵Hb血症還可見於一種HbM遺傳性高鐵Hb血症。這種疾病是由於a58組→酪突變,酪氨酸占據了血紅素Fe原子的配基位置,使之呈現穩定的高鐵狀態,患者有紫紺症狀和繼發性紅細胞增多。
(4)Hb與氧的親和力異常增加:見於輸入大量庫存血液或鹼性液體,也見於某些血紅蛋白病。庫存血液的紅細胞內2,3-DPG含量低使氧合血紅蛋白解離曲線左移;基因的突變,a鏈第92位精氨酸被亮氨酸取代時,Hb與O2的親和力比正常高几倍。
2、血氧變化的特點
貧血引起缺氧時,由於外呼吸功能正常,所以PaO2、SaO2正常,但因Hb數量減少或性質改變,使氧容量降低導致CaO2減少。
CO中毒時,其血氧變化與貧血的變化基本是一致的。但是CO2max在體外檢測時可以是正常的,這因在體外用氧氣對血樣本進行了充分平衡,此時O2已完全競爭取代HbCO中的CO形成氧合Hb,所以血CO2max可以是正常的。
血液性缺氧時,血液流經毛細血管時,因血中HbO2總量不足和PO2下降較快,使氧的彌散動力和速度也很快降低,故A-VdO2低於正常。
Hb與O2親和力增加引起的血液性缺氧較特殊,其PaO2正常;CaO2和SaO2正常,由於Hb與O2親和力較大,故結合的氧不易釋放導致組織缺氧,所以PvO2升高;CvO2和SvO2升高,A-VdO2小於正常。
3、皮膚、黏膜顏色變化
單純Hb減少時,因氧合血紅蛋白減少,另外患者毛細血管中還原Hb未達到出現紫紺的閾值,所以皮膚、黏膜顏色較為蒼白;HbCO本身具有特別鮮紅的顏色,CO中毒患者時,由於血液中HbCO增多,所以皮膚、黏膜呈現櫻桃紅色,嚴重缺氧時由於皮膚血管收縮,皮膚、黏膜呈蒼白色;高鐵Hb血症時,由於血中高鐵Hb含量增加,所以患者皮膚、黏膜出現深咖啡色或青紫色;單純的由Hb與O2親和力增高時,由於毛細血管中脫氧Hb量少於正常,所以患者皮膚、黏膜無紫紺。
循環性缺氧
循環性缺氧(circulatoryhypoxia)指組織血流量減少使組織氧供應減少所引起的缺氧,又稱為低動力性缺氧(hypokinetichypoxia)。循環性缺氧還可以分為缺血性缺氧(ischemichypoxia)和淤血性缺氧(congestivehypoxia)。缺血性缺氧是由於動脈供血不足所致;淤血性缺氧是由於靜脈回流受阻所致。
1、原因
 缺氧患者
缺氧患者循環性缺氧的原因是血流量減少,血流量減少可以分為全身性和局部性二種。
(1)全身性血流量減少
(2)局部性血流量減少
2、血氧變化的特點
單純性循環障礙時,血氧容量正常;PaO2正常、CaO2正常、SaO2正常。由於血流緩慢,血液流經毛細血管的時間延長,使單位容積血液彌散到組織氧量增加,CvO2降低,所以A-VdO2血氧差也加大;但是單位時間內彌散到組織、細胞的氧量減少,還是引起組織缺氧。局部性循環性缺氧時,血氧變化可以基本正常。
3、皮膚、黏膜顏色變化
由於靜脈血的CvO2和PvO2較低,毛細血管中脫氧Hb可超過50g/L,可引發皮膚、黏膜紫紺。
組織性缺氧
組織性缺氧(histogenoushypoxia)是指由於組織、細胞利用氧障礙所引起的缺氧。
1、原因
 呼吸系統
呼吸系統(1)抑制細胞氧化磷酸化
細胞色素分子中的鐵通過可逆性氧化還原反應進行電子傳遞,這是細胞氧化磷酸化的關鍵步驟。以氰化物(cyanide)為例,當各種無機或有機氰化物如:HCN、KCN、NaCN、NH4CN和氫氰酸有機衍生物(多存在於杏、桃和李的核仁中)等經消化道、呼吸道、皮膚進入體內,CN-可以迅速與細胞內氧化型細胞色素氧化酶三價鐵結合形成氰化高鐵細胞色素氧化酶(CNˉ+Cytaa3Fe3+→Cytaa3Fe3+-CNˉ),失去了接受電子能力,使呼吸鏈中斷,導致組織細胞利用氧障礙。0.06gHCN可以導致人的死亡。高濃度CO也能與氧化型細胞色素氧化酶aa的Fe2+結合,阻斷呼吸鏈。硫化氫、砷化物和甲醇等中毒是通過抑制細胞色素氧化酶活性而阻止細胞的氧化過程。抗黴菌素A和苯乙雙胍等能抑制電子從細胞色素b向細胞色素c的傳遞,阻斷呼吸鏈導致組織中毒性缺氧。
(2)線粒體損傷
引起線粒體損傷的原因有:強輻射、細菌毒素、熱射病、尿毒症等。線粒體損傷,可以導致組織細胞利用氧障礙和ATP生成減少。
(3)呼吸酶合成障礙
維生素B1、B2、尼克醯胺等是機體能量代謝中輔酶的輔助因子,這些維生素缺乏導致組織細胞對氧利用和ATP生成發生障礙。
2、血氧變化的特點
組織性缺氧時,血氧容量正常,PaO2、CaO2、SaO2一般均正常。由於組織細胞利用氧障礙(內呼吸障礙),所以PvO2、CvO2、SvO2增高,(A-V)dO2小於正常。患者的皮膚、黏膜顏色因毛細血管內氧合Hb的量高於正常,故常呈現鮮紅色或玫瑰紅色。
臨床常見的缺氧多為混合性缺氧。例如肺源性心臟病時由於肺功能障礙可引起呼吸性缺氧,心功能不全可出現循環性缺氧。
功能變化
機體吸入氧,並通過血液運輸到達組織,最終被細胞所感受和利用。因此,缺氧的本質是細胞對低氧狀態的一種反應和適應性改變。當急性嚴重缺氧時細胞變化以線粒體能量代謝障礙為主(包括組織中毒性缺氧);慢性輕度缺氧細胞以氧感受器的代償性調節為主。
 呼吸系統
呼吸系統1、缺氧時細胞能量代謝變化
(1)無氧酵解增強:當PaO2降低時,線粒體周圍的PO2低於0.04~0.07kPa時,氧作為有氧氧化過程的最終的電子接受者出現缺額,線粒體的有氧代謝發生障礙,ATP生成減少,胞漿內ADP增加。胞漿內ADP增高可使磷酸果糖激酶、糖酵解過程加強,並在一定的程度上可補償細胞的能量不足,但酸性產物增加。
(2)利用氧的能力增強:長期慢性和輕度缺氧時,細胞內線粒體數量增多,生物氧化還原酶(如琥珀酸脫氫酶、細胞色素氧化酶)活性增強和含量增多,使細胞利用氧的能力增強。
2、細胞的氧敏感調節與適應性變化
(1)化學感受器興奮
(2)血紅素蛋白(hemeprotein)感受調節:血色素蛋白是指含有卟啉環配體的一類蛋白質,如血紅蛋白、細胞色素aa3、P450、含細胞色素b558的輔酶Ⅱ(NADPH)氧化酶等。感受調節方式有兩種:
①構象改變當O2結合於血紅素分子中央的Fe2+,引起Fe2+轉位到卟啉環平面上,反之相反。這種構象的變化可能影響血紅素蛋白的功能。例如:CO與氧化型細胞色素氧化酶aa的Fe2+結合,使氧化型細胞色素氧化酶失去了傳遞電子的作用。
②信使分子NADPH氧化酶可與細胞周圍環境中O2結合,並把O2轉變為O2-,再生成H2O2。H2O2經過Feton反應轉變為羥自由基(OH-)進行氧信號的傳導。正常時,細胞內H2O2濃度相對較高,抑制低氧敏感基因的表達。低氧時,細胞內H2O2和OH-生成減少,還原型谷光甘肽(GSH)氧化轉變成氧化型谷光甘肽(GSSG)受到抑制,導致某些蛋白巰基還原型增加,從而使一些轉錄因子的構象發生改變,促進低氧敏感基因的轉錄表達。
3、HIF-1感受調節
研究認為,HIF-1(hypoxiainducedfactor-1)是受控於氧濃度變化的一個至關重要的轉錄因子。細胞核內HIF-1作為低氧敏感基因的啟動子與靶基因的低氧反應元件(HRE,5-RCGTG-3)結合,啟動基因轉錄和蛋白質翻譯。
4、紅細胞適應性增多
在高原居住的人和長期慢性缺氧的人,紅細胞可以增加到6×106/㎜3,Hb達21g/dl。其增加機制是,當缺氧時,低氧血可以刺激近球細胞,使其生成促紅細胞生成素(erythropoiesis-stimulatingfactor,EPO)增加。EPO可以刺激RBC系單向幹細胞分化為原RBC和增殖、成熟。另外。EPO可促使Hb合成和網織紅細胞進入血液,血中紅細胞和Hb增加,提高了血液中血氧容量。最終提高了血液攜帶氧的能力使氧含量增加,從而增強對組織器官的O2供應。
5、肌紅蛋白(Mb)增加
由於Mb與氧的親和力比Hb的大,如氧分壓降為10mmHg時,Hb的氧飽和度約為10%,而Mb的氧飽和度可達70%,因此,當運動員進行劇烈運動使肌組織氧分壓進一步降低時,Mb可釋放出大量的氧供組織、細胞利用。Mb增加可能具有儲存氧的作用。
細胞損傷
缺氧性細胞損傷(hypoxiccelldamage)常為嚴重缺氧時出現的一種失代償性變化。其主要表現為細胞膜、線粒體及溶酶體的損傷。
 缺氧患者
缺氧患者1、細胞膜變化
細胞膜電位降低常先於細胞內ATP含量的減少,膜電位降低的原因為細胞膜對離子的通透性增高,導致離子順濃度差通過細胞膜,繼而出現鈉內流、鉀外流、鈣內流和細胞水腫等一系列改變。
(1)Na+內流:使細胞內Na+濃度增多並激活Na+-K+泵,在泵出胞內Na+同時又過多消耗ATP,ATP消耗又將促進線粒體氧化磷酸化過程和加重細胞缺氧。細胞內Na+濃度過高必然伴隨水進入胞內增加引起細胞水腫。細胞水腫是線粒體、溶酶體腫脹的基礎。
(2)K+外流:由於Na+-K+泵功能障礙,細胞外K+不能被泵到胞漿內,細胞內缺K+導致合成代謝障礙,各種酶的生成減少並進一步影響ATP的生成和離子泵的功能。
(3)Ca2+內流:細胞內外Ca2+濃度相差約1000倍,細胞內低Ca2+濃度的維持依賴膜上Ca2+泵功能。嚴重缺氧時,由於ATP生成減少,膜上Ca2+泵功能降低,胞漿內Ca2+外流和肌漿網攝取Ca2+障礙,使胞漿內Ca2+濃度增高。細胞內Ca2+增多並進入線粒體內抑制了呼吸鏈功能;Ca2+和鈣調蛋白(calmodulin)激活磷脂酶,使膜磷脂分解,引起溶酶體損傷及其水解酶的釋放,細胞自溶;胞漿內Ca2+濃度過高可以使黃嘌呤脫氫酶轉變為黃嘌呤氧化酶,增加自由基形成,加重細胞損傷。
2、線粒體的變化
缺氧可損傷線粒體,線粒體損傷又可導致缺氧,兩者互為因果。缺氧引起線粒體受損的原因是嚴重缺氧可明顯抑制線粒體呼吸功能和氧化磷酸化過程,使ATP生成更減少;持續較長時間嚴重缺氧,可以使線粒體的基質顆粒減少或消失,基質電子密度增加,脊內腔擴張,脊腫脹、崩解,外膜破裂等。
3、溶酶體的變化
缺氧時因糖酵解增強使乳酸生成增多和脂肪氧化不全使酮體增多,導致酸中毒。pH降低和胞漿內鈣增加使磷脂酶活性增高,使溶酶體膜的磷脂被分解,膜通透性增高,溶酶體腫脹、破裂和釋出大量溶酶體酶,進而導致細胞及其周圍組織的溶解、壞死。細胞內水腫、自由基的作用也參加溶酶體損傷機制。
代謝變化
缺氧對器官的影響,取決於缺氧發生的程度、速度持續時間和機體的功能代謝狀態。慢性輕度缺氧主要引起器官代償性反應;急性嚴重的缺氧,器官常出現代償不全和功能障礙,甚至引起重要器官產生不可逆損傷,導致機體的死亡。
 缺氧症狀
缺氧症狀呼吸系統的變化
(一)代償性反應
1、呼吸加深加快
2、胸廓呼吸運動增加
主要是低氧血症引起的呼吸運動增加使胸內負壓增大,促進了靜脈回流增加,增加心輸出量和肺血流量,有利於氧的攝取和運輸。
低張性缺氧所引起的肺通氣變化與缺氧持續的時間有關。
4000m高原的空氣PO2為100mmHg,肺泡氣PO2為55mmHg左右。因此,在化學感受器的低氧感受下,肺通氣量立即增加,由於空氣稀薄,PCO2也低,CO2呼出增加(發生呼吸性鹼中毒和低氧血症),PaCO2降低,減低了對延髓的中樞化學感受器的刺激,限制肺的通氣量增加,所以,早期肺通氣量只比海平面高65%;數日後,通過腎代償性排除HCO3-,腦脊液內的HCO3-也通過血腦屏障進入血液使腦組織中pH逐漸恢復正常,對延髓的中樞化學感受器的刺激抑制逐漸解除,肺的通氣量可增加至海平面的5-7倍;長期居住者肺通氣量逐漸回落,至僅比海平面高15%,這可能與外周化學感受器對低氧的敏感性降低有關。這也是一種慢性適應過程,因為肺通氣每增加1L,呼吸肌耗氧增加0.5ml,所以長期呼吸運動增加顯然對機體不利。
(二)呼吸功能障礙
高原肺水腫(highaltitudepulmonaryedema,HAPE),表現為呼吸困難、咳嗽、血性泡沫痰、肺部有濕性羅音,皮膚黏膜發紺等。其發病機制與以下因素有關:
①缺氧引起外周血管收縮,回心血量增加和肺血量增多,加上缺氧性肺血管收縮反應使肺血流阻力增加,導致肺動脈高壓。
②肺血管收縮強度不一使肺血流分布不均,在肺血管收縮較輕或不發生收縮的部位,肺泡毛細血管血流增加、流體靜壓增高,引起壓力性肺水腫。
③肺內血壓高和流速快對微血管的切應力(流動血液作用於血管的力在管壁平行方向的分力)增高。
④肺的微血管壁通透性增高,例如,補體C3a、LTB4和TXB2等血管活性物質可能導致微血管內皮細胞損傷和通透性增高。
肺水腫影響肺的換氣功能,可使PaO2進一步下降,加重缺氧。PaO2過低可直接抑制呼吸中樞,使呼吸抑制,肺通氣量減少,導致呼吸衰竭。
循環系統的變化
 缺氧急救
缺氧急救1、心輸出量增加
導致心輸出量增加的主要機制是:
①心率加快:當吸入含8%O2的空氣時,心率可增加一倍。目前認為,心率加快很可能是通氣增加所至肺膨脹對肺牽張感受器的刺激,反射性抑制迷走神經對心臟的效應;但呼吸運動過深產生過度牽張刺激使心率減慢和血壓下降。
②心肌收縮性增強:缺氧作為一種應激原,可使交感神經興奮和兒茶酚胺釋放增多,作用心臟β-腎上腺素能受體,使心率加快,心肌收縮性增強。
③靜脈回流增加:缺氧時胸廓運動和心臟活動增強,胸腔內負壓增大,靜脈回流增加和心輸出量增加。
2、血液重分布
急性缺氧時,皮膚、腹腔內臟因交感神經興奮,縮血管作用占優勢,使血管收縮;而腦血管收縮不明顯;冠脈血管在局部代謝產物(如CO2、H+、K+、磷酸鹽、腺苷及PGI2等)的擴血管作用下血流增加。這種全身性血流分布的改變,顯然對於保證生命重要器官氧的供應是有利的。
3、肺血管收縮(肺血管對缺氧的反應與體血管相反)
①交感神經興奮作用使肺血管收縮急性缺氧時所致交感神經興奮性可作用於肺血管的α1受體引起血管收縮反應。慢性低氧時肺內血管平滑肌出現受體分布的改變:α1受體增加,β受體密度降低,導致肺血管收縮增強。
②體液因子的作用使肺血管收縮。肺組織內肥大細胞、肺泡巨噬細胞、血管內皮細胞以及血管平滑肌細胞等能釋放各種血管活性物質,如:肥大細胞脫顆粒釋放組胺、VEC釋放PGI2、ET增加引起肺血管收縮。在血管收縮過程中,縮血管物質增加起主導作用,擴血管物質的增加起反饋調節作用。
③血管平滑肌對低氧的直接感受。正如缺氧時細胞的代謝和功能變化一節所述,缺氧可直接通過肺血管平滑肌細胞膜上對氧敏感的鉀通道關閉,使細胞內K+外流減少,膜電位下降,細胞興奮性增高、極化加速和細胞外Ca2+內流增強,最終導致了肺血管收縮。
慢性缺氧除了肺血管收縮導致肺動脈高壓外,還有肺內血管壁中層平滑肌肥大、增厚以及彈力纖維和膠原纖維增生使血管的管徑變小、血流阻力增加。
4、毛細血管增生
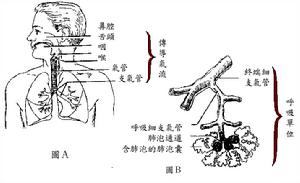 呼吸系統
呼吸系統組織細胞的長期輕度缺氧,可通過HIF-1a的低氧感受使細胞合成與釋放VGEF增多,毛細血管在缺氧的組織增生(見缺氧時細胞的代謝和功能變化)。這種現象在腦、肥大的心肌、實體腫瘤和骨骼肌中,毛細血管增生更加顯著。
血液系統的變化
缺氧可使骨髓造血增強和氧合血紅蛋白解離曲線右移。
1、紅細胞增多
2、氧合血紅蛋白解離曲線右移
缺氧時,紅細胞內2,3-DPG增加,導致氧合Hb解離曲線右移,Hb易將結合的氧釋放出供組織利用。
(1)紅細胞內生成2,3-DPG增多的原因有兩個方面:
①低張性缺氧時氧合Hb減少,脫氧Hb增多,前者中央穴孔小,不能結合2,3-DPG;後者中央孔穴較大,可結合2,3-DPG。當脫氧Hb增多時,紅細胞內游離的2,3-DPG減少,2,3-DPG對磷酸果糖激酶及二磷酸甘油變位酶(diphosphoglyceratemutase,DPGM)的抑制作用減弱,從而使糖酵解增強,2,3-DPG生成增多。
②低張性缺氧因代償性肺過度通氣引起呼吸性鹼中毒,以及缺氧時紅細胞記憶體在的大量脫氧Hb稍偏鹼性,使紅細胞內pH增高,從而激活磷酸果糖激酶和抑制2,3-DPG磷酸酶(2,3-DPGphosphatase,2,3-DPGP)活性。前者使糖酵解增強,2,3-DPG合成增加;後者使2,3-DPG的分解減少。
(2)2,3-DPG增多使氧合Hb解離曲線右移的機制是:
① 與2,3-DPG結合的脫氧Hb其空間構型較為穩定,不易於氧結合;
② 2,3-DPG是一種不能透出紅細胞的有機酸,其增多可降低紅細胞內pH,pH下降通過Bohr效應使氧合Hb解離曲線右移。但是,當PaO2低於8kPa時,氧離曲線右移可明顯影響肺部血液對氧的攝取。
3、血紅蛋白表型重建
中樞神經系統的變化
中樞神經系統是對缺氧最為敏感的器官,因為腦對氧的需求非常高。腦重量僅為體重的2%,而腦血流占心輸出量15%,腦耗氧量占總耗氧量23%,所以,腦對缺氧十分敏感,臨床上腦完全缺氧5-8min後可發生不可逆的損傷。
急性缺氧可引起頭痛、情緒激動,思維力、記憶力、判斷力下降或喪失以及運動不協調等。嚴重缺氧可使腦組織發生細胞腫脹、變性、壞死及腦間質水腫等形態學變化,這與缺氧及酸中毒使腦微血管通透性增高引起腦間質水腫有關。這些損傷常常在缺氧幾分鐘內發生。且不可逆。腦血管擴張、腦細胞及腦間質水腫可使顱內壓增高,由此引起頭痛、嘔吐、煩躁不安、驚厥、昏迷,甚至死亡。慢性缺氧則易疲勞、嗜睡、注意力不集中等症狀。
極嚴重缺氧可導致昏迷、死亡的發生機制是由於神經細胞膜電位降低,神經遞質合成減少;腦細胞能量代謝障礙,ATP減少,細胞膜通透性增加;酸中毒,細胞內游離Ca2+增多,溶酶體酶的釋放以及細胞水腫等因素導致引起中樞神經系統功能障礙。
所謂高原腦水腫(highaltitudecerebraledema,HACE)發病機制除了缺氧引起腦血管擴張、腦血流增多外,可能還與下列因素有關。1)腦細胞水腫;2)血腦屏障功能受損,3)腦靜脈內血栓形成,進一步加重腦水腫形成。
