概述
 Leber遺傳性視神經病變
Leber遺傳性視神經病變流行病學
發病多在20歲左右,少數可發生於10歲以下或30歲以上最小可在5歲最大可在70歲。根據的經驗國人發病有偏低的趨勢,15歲以下約占1/4。根據文獻統計,男性與女性患者之比為5∶1~9∶1,西方患病率男與女比為3∶1或9∶1,中國男女之比為57%∶43%即約為6∶4,與日本國相近似,顯示黃種人女性發病有增多現象。LHON在北歐及東方亞洲人群發病率高。兩眼同時或先後發病,可間隔數天或數年,也可延長1年雙眼受累,單眼發病罕見,多呈急性亞急性發作其後呈慢性逐漸發展。
病因
 線粒體DNA
線粒體DNA發病機制
LHON繼發位點有增多的趨勢,已達22個,雖然它們在LHON患者中發生率比對照組中高但在對照家系中也存在,一般認為可能為多態性,母親發現有異質性,且單獨存在時不患病。有人認為繼發位點不能改變該病的表現,但也有人認為可增強表型表達的可能性可起媒介作用或有低危險性。
臨床表現
 神經纖維
神經纖維1.顯性遺傳性視神經萎縮(dominant optic atrophy) 較為少見是一種視神經的生活力缺失(abiotrophy)。多發生於10歲以前多數在4~6歲開始發生雙眼中等程度的視力障礙,大約40%的病人視力在0.3以上,僅15%視力損害較重,低於0.1以下。據統計未見有視力降至手動及光覺者。
外眼及眼前節正常。眼底表現為視盤顳側輕微蒼白,少數視力障礙嚴重者可伴有眼球震顫。視野檢查可查見中心、旁中心或啞鈴型暗點白色周圍視野正常,但因病人有藍色盲,因此藍色周圍視野反較紅色視野為小。利用圖形及閃光VEP檢查,可查出病人的VEP振幅較低,峰潛時延長
2.隱性遺傳性視神經萎縮(recessive optic atrophy) 更為罕見,多在出生後或3~4歲以前發病因此又稱為先天性隱性遺傳性視神經萎縮。半數以上的病人父母有血緣關係。病人視力多有嚴重損害或完全失明,並有眼球震顫。如果能查視野,可見視野縮小及旁中心暗點眼底表現視神經全部萎縮、凹陷和視網膜血管變細。因此有時易與毯層視網膜變性相混淆,但ERG可作鑑別:LEBER遺傳性視神經病變ERG正常,而毯層視網膜變性者ERG熄滅
LHON家系成員可表現有其他的神經異常,如外周神經病變、頭痛、偏頭痛、智力障礙、震顫、癲癇、耳聾脊髓後柱受累、小腦性共濟失調運動失調、肌張力障礙、膀胱無力征等,其他尚可見心臟傳導障礙,多發生在11778位點突變;3460位點突變者易患預激綜合徵在LHON家系中可見有類似多發性硬化的脫髓鞘疾病。臨床上有報導在發生視神經病變的同時可見與多發性硬化(MS)相符的症狀和體徵。這些患者的腦脊液及磁共振成像檢查可發現多發性硬化的典型表現,人群調查中未顯示在多發性硬化患者中mtDNA突變發生率有所增加看來該兩病之間並不一定有聯繫,但可合併發生。MS患者如合併LHON位點突變,其視神經炎的預後會更差尚可見有些合併嚴重神經系統異常的LHON,被稱為Leber綜合徵。如視神經病變尚有運動失調、痙攣、精神障礙骨骼肌異常、急性嬰兒腦病發作。
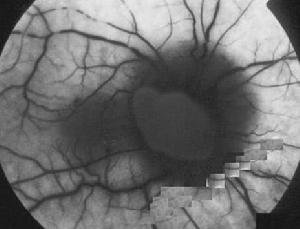
有人認為該病原發於視網膜血管的改變而稱為“毛細血管擴張性微血管病變”,此種微血管病變在發病前即可出現因此對該病家族成員應仔細地追蹤檢查眼底Smith認為早期可有視盤周圍毛細血管擴張性微動脈血管改變,視盤周圍視網膜神經纖維層腫脹和視盤無滲漏三聯征。慢性期則逐漸視盤色淡甚至蒼白。
併發症 多發展為視神經萎縮也可見急性球後視神經炎。
診斷 主要根據病史、臨床表現以及mtDNA實驗室檢查結果判定診斷。
檢查
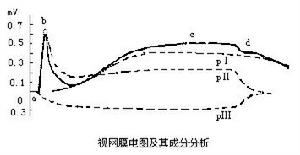 視網膜電圖
視網膜電圖1.眼底螢光血管造影(FFA) 在急性期視盤呈強螢光,血管高度擴張,視盤黃斑束毛細血管充盈、延緩缺損等,FFA檢查可早期發現有發病可能的患者及攜帶者,因此可用於遺傳諮詢對無症狀而有輕微血管改變者,有可能在數年後發病。
2.腦誘發電位(EPS)以及頭顱CT,MRI檢查 部分病例可發現與多發性硬化相符的情況。
3.視網膜電圖(ERG)檢查 可用於鑑別診斷。
治療
LEBER遺傳性視神經病變至今尚無有效的治療方法。因為有些病人在病程中視力可以自然恢復,所以對任何治療效果的評價均應慎重。此病被作為基因治療的重點研究對象之一。
為減少對視神經的毒性損害,應告誡患者戒菸和戒酒。雖然臨床有使用神經營養藥物治療,但並無肯定的療效。日本對於急性期病例使用血管擴張劑艾地苯醌(idebenone)聯合維生素B2、維生素C、泛癸利酮(輔酶Q10)和前列腺素類的降眼壓藥異丙烏諾前列酮(Isopropyl unoprostone),旨在縮短視力恢復的時間。
預後
有些Leber遺傳性視神經病變病人,在視力減退數月甚至數年以後,視力可以部分甚至全部恢復。這種視力部分或全部恢復的病例,據統計可分別為29%及12%。視力一旦有所恢復通常很少會再減退。
預防
戒菸、戒酒或許有效,對女性患者如已證實為女性攜帶者應進行產前檢查,以利優生。
