定義
 艾森曼格綜合症
艾森曼格綜合症艾森曼格複合病一詞,以往曾用以稱一種複合的先天性心臟血管畸形,包括室間隔缺損、主動脈右位、右心室肥大而肺動脈正常或擴大者。病人有紫紺。本病與法樂四聯症不同之處在於並無肺動脈口狹窄。自心臟導管檢查在臨床上廣泛套用以來,通過對先心病的血流動力學研究,艾森曼格綜合徵多用以指心室間隔缺損合併肺動脈顯著高壓伴有右至左分流的病人。推而廣之,心房間隔缺損、動脈導管未閉、主動脈-肺動脈間隔缺損等先心病發生肺動脈顯著高壓而有右至左分流時,都可有類似的臨床表現,亦可以歸入本綜合徵的範疇。因此本綜合徵可以稱為肺動脈高壓性右至左分流綜合徵。
概述
 艾森曼格綜合症
艾森曼格綜合症在1958年,Paul Wood描述這一複合征。通過一些不同類型病例包括心臟和大動脈的缺損,這些都導致肺動脈高壓伴隨肺血管阻力上升(>800dynes.s/cm5)。這一複合征於是被重命名為艾森曼格綜合徵。並被定義為系統水平的肺動脈高壓。由於肺血管阻力上升伴隨反響或雙向分流,通過任何巨大的體肺分流例如一個病人伴隨動脈導管未閉,也包含在這一定義內。這一新定義含有不可逆性或無法手術的這么一類有先天性心臟疾病的患者。
今天儘管我們對艾征的病理生理方面的理解和對此病的治療已有了許多進步。但仍沒有治療措施能阻斷此病的進展。一旦確診為該病,除了進行肺臟移植或心肺聯合移植。此病伴隨很高的患病率和死亡率。
臨床表現
 艾森曼格綜合症
艾森曼格綜合症艾森曼格征是指由於肺血管主力升高導致肺動脈壓力上升,伴隨雙向或反向分流通過一個大的心內或心外的先天性心臟缺損。這是經由未被矯正的先天缺損體肺分流的結果。有幾種先天性缺損伴隨高風險的肺血管疾病,如室缺,動脈導管未閉,房室通道缺損。近三分之一的未行先心病矯治的病人,即指未做心臟手術或心臟移植或死於非心源性誘因,這些病人中的三廢紙一死於肺血管疾病。
及時診斷巨大的體肺分流是阻止艾征的關鍵。在生後的頭兩年閉合缺損。不大可能導致肺血管阻塞性疾病.嬰幼兒有巨大的非限制性的缺損,通常早期有充血性的心衰症狀;也就是說體弱,多汗,呼吸急促,這是由於從左向右分流穿過心臟缺損上升的肺血流量引起的。持續暴露於從左向右分流的高切變應激多年之後,相對體循環而言,肺血管阻力的上升導致雙向的分流,或右向左分流;紫紺緊隨著出現,也可以很嚴重。在很少病例沒有充血性心衰史或嬰兒期的體弱多病。在這些孩子中反向分流可能,在這些在出生頭兩年。
自然病程
 艾森曼格綜合症
艾森曼格綜合症艾征自然病程有一個很大的跨度,儘管總的來講,其生存率對於原發性肺動脈高壓。大多數病人能生存到三到四個十年,而且儘管非常少但仍有報導個別病人生存到第七個十年。
當同時合併一個潛在的染色體畸形如21三體綜合症,這種病人經常比沒有染色體畸形的病例生存的時間短。在艾征作為一個主要的臨床基本概念出現後,先天性心臟病的自然病史研究開始實施。第二次自然病史研究(1993)繼1958~1969第一次疾病自然史研究(1993)支持艾征病人自其被診斷後仍有生存幾個十年;及54%的病人(在98個未手術的室缺病人+艾征)的病人在艾征被診斷後仍存活20年。
Clarson等在以前的研究中也支持這一觀點。他們認為10~19歲艾征病人在被診斷後5年存活率95%;年齡超過20歲者5年存活率為56%。
目前儘管唯一的治療這種疾病措施是肺移植或心肺聯合移植;在過去的10年間幾項進步已能促使病人獲得更高的生活質量伴隨著活動能力的提高。
輔助診斷
除上述臨床症狀與體徵外,還可進行進一步檢查:
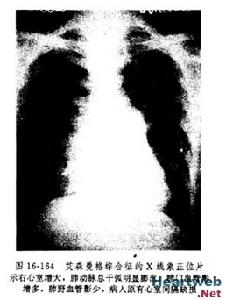
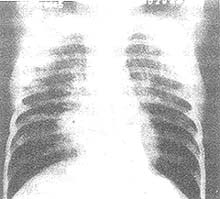
1、X線診斷心臟增大,左、右室均增大,肺動脈段凸出,肺門血管影增粗,搏動明顯,周圍血管影則纖細,此為肺動脈高壓的典型表現。
2、心電圖檢查常呈現右心室肥厚,可同時伴有右心房肥大,有些病例左室亦可肥厚。
3、心導管檢查肺動脈壓力明顯增高,與主動脈壓相等或更高,動脈血氧飽和度降低。室間隔缺損者,右心室血氧可高於右心房,說明在心室水平有雙向分流,導管可通過缺損直接進入左室和主動脈。動脈導管未閉者,肺動脈血氧高於右心室,說明在肺動脈水平有雙向分流。下肢血氧飽和度降低較上肢為明顯,導管可通過未閉的動脈導管而進入降主動脈。
4、心血管造影套用選擇性心血管造影可確定缺損部位,如為動脈導管未閉,造影劑通過未閉的動脈導管進入降主動脈,主動脈與肺動脈同時顯影。如為室間隔缺損,則左心室、主動脈與右心室同時顯影。
病理生理
 艾森曼格綜合症
艾森曼格綜合症在艾征病人,這種病理改變會因為通過上升,肺血管流量引起外周肌伸展從不同外周細胞和中間細胞在毛細血管前血管與肺血管內皮細胞的損害一樣,這將導致一系列細胞水平的變化(涉及肺血管疾病的病原學)。例如環鳥苷酸和環腺苷酸,被變化的非內皮依賴性物質激活(異丙級腎上腺素、硝普鈉)和內皮依賴性物質(乙醯膽鹼)機制導致肺血管擴張。
伴隨內皮的功能障礙,肺血管阻力上升並最終導致左向右分流減少。最終閉合當阻力顯著提高,分流變為雙向的,接著變為右向左。長遠的看,有肺血管阻力上升的病人進行了缺損的病人情況比有同樣損害但為進行修補的病人情況要差。
這可能與缺乏一種在左室與右室循環之間交通,而這種交通在處於肺動脈高壓風險中的病人起一種“壓力安全閥”作用。 為支持這一理論,以前研究證實艾征病人以右向左分流,也就是血液不飽和為代價來保持正常右房壓。
另外,艾征病人的心輸出量比原發肺動脈高壓要大,從推測上講是繼發於壓力安全閥的機理存在而出現的。
治療
 艾森曼格綜合症
艾森曼格綜合症睡眠過程中吸氧能延緩艾森曼格綜合徵患者紅細胞增多症的進展;
患者運動時氧需求量增加,而血液輸送氧的能力不能相應增加,因此運動時吸氧對患者有益;
右心衰患者安靜時對氧需求也較大,也應給予吸氧治療;
在急性呼吸道感染時吸氧能防止低氧血症進一步加重。
2、洋地黃類藥物治療艾森曼格綜合徵
儘管對右心衰患者是否套用強心藥仍存在爭議,但有報導表明對肺動脈高壓合併右 心衰患者套用洋地黃類藥物可增加心輸出量。對缺氧患者聯合套用洋地黃和利尿劑應特別注意維持電解質的平衡,因為電解質紊亂會增加洋地黃中毒的危險。
3、利尿劑在艾森曼格綜合徵治療中的套用
對艾森曼格綜合徵合併右心衰竭的患者套用利尿劑可緩解肝臟淤血,減少血容量。 但要避免利尿過度,因為有些患者需要一定前負荷才能維持有效心輸出量。對紅細胞顯著增多的患者應警惕。利尿後血液粘滯度升高而增加中風或其他併發症的危險。
4、艾森曼格綜合徵抗凝治療
艾森曼格綜合徵由於嚴重缺氧可導致紅細胞明顯增多,並且活動受限導致血流不 暢、右室擴大及肺血流緩慢等極易並發血栓栓塞,因而抗凝劑治療可提高艾森曼格綜合徵患者的生存率、運動耐力和生活質量。
