病因
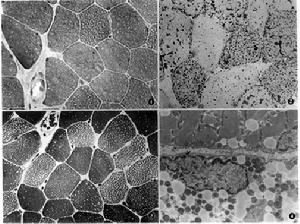 脂質沉積性肌病
脂質沉積性肌病本病半數患者有家族史屬常染色體隱性遺傳。人體脂肪代謝是全身性的脂肪代謝障礙可以發生在整個機體並成為家族遺傳性疾病。例如脂質沉積在腦組織可引起腦脂質沉積症,表現為腦病綜合徵,此型疾病主要見於嬰幼兒並常伴以心臟、肝臟等內臟損害。脂質沉積性肌病是脂肪代謝障礙累及骨骼肌的一種表現脂肪代謝生化轉變過程中的任何環節出現障礙均可導致脂質在肌肉或全身各器官內堆積致病。引起LSM的病因常見者為肉鹼缺乏或肉鹼棕櫚醯基轉移酶缺乏。
發病機制:
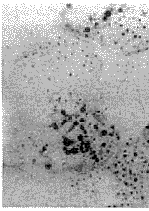 骨骼肌
骨骼肌正常骨骼肌在靜止和運動時,其能量均來源於脂肪酸在線粒體內的β-氧化作用,體內的脂肪酸根據所含C原子的多少,分為短鏈(2~4C)、中長鏈(4~12C)及長鏈(12C以上)三種肉鹼是人體重要的代謝活性物質98%儲存在肌肉內,其餘見於肝腎及細胞外液中。肉鹼存在於線粒體內外膜之間它有兩個基本作用:首先是將長鏈脂肪酸轉移而穿過線粒體內膜,進入基質進行β-氧化;其次是通過調節線粒體內輔酶A(CoA)和脂醯輔酶A(acyl-CoA)的比值(CoA/acyl-CoA),防止acyl-CoA線上粒體內集聚從而維護膜的穩定性。長鏈脂肪酸的氧化過程是一系列生物化學轉變過程脂肪酸與CoA線上粒體外膜上脂醯CoA合成酶(AS)的催化作用下形成高能硫脂鍵,即活化的脂醯CoA,後者並不能直接穿過線粒體內膜,而必須依靠位於線粒體外膜內側面上的肉鹼棕櫚醯基轉移酶Ⅰ(carnitinepalmtoyltransferaseCPTⅠ)的作用將脂醯CoA和肉鹼轉變為脂醯肉鹼(acyl-carnitine)後,在肉鹼-脂醯肉鹼轉位酶(carnitineacylcarnitinetranslocase,CT)的催化作用下才能通過線粒體內膜進入基質脂醯肉鹼進入基質後,又須通過位於線粒體內膜內側面的肉鹼棕櫚醯基轉移酶Ⅱ(CPTⅡ)的作用,將脂醯肉鹼轉換為脂醯CoA和肉鹼前者在基質中進行β-氧化,而肉鹼則再次通過酶的作用流出線粒體內膜以便再次以同樣的方式,使脂醯CoA穿過線粒體內膜。因此肉鹼成為長鏈脂肪酸進入線粒體基質氧化的運載工具。這種循環過程稱作肉鹼環(carnitinecycle)。
人體肉鹼75%來源於食物它富含於紅肉和魚類由腸上皮細胞緩慢吸收,然後進入肝臟。肉鹼經糞及尿液排出體外有些則再現於膽汁,正常飲食不能滿足整個機體的需要而須內源性合成其原料為賴氨酸和甲基賴氨酸,並主要在肝內合成。
 線粒體
線粒體病理特點:多採取肌肉活檢標本,冰凍切片經組織化學染色法進行觀察。不論是肉鹼缺乏還是肉鹼棕櫚醯基轉移酶缺乏的患者,光鏡下均可發現:HE及改良Gomori三染色顯示肌質內和肌膜下大量散在大小不等的圓形空泡或缺損,油紅O染色見該處為脂滴顆粒。ATP酶染色提示脂質在Ⅰ型肌纖維內沉積最多,其次為ⅡA型再次為ⅡB型肌纖維。其原因可能和Ⅰ型肌纖維更要依賴脂肪代謝有關Ⅰ型肉鹼棕櫚醯基轉移酶缺乏的患者在肌紅蛋白尿發作期間,可見肌纖維壞死且以Ⅰ型肌纖維損害較重,並可繼以再生。
電鏡觀察可見脂滴直徑大小不一,約在不足1微米至數個微米之間,脂滴無膜平行分布於肌原纖維間或在肌膜下堆積,但應注意不同病人隨不同病程其含量變異較大線粒體數目及大小均增加,其嵴不清晰由於肌肉的脂質和葡萄糖代謝均線上粒體內進行,故在LSM患者的肌纖維中有時可見糖原顆粒同時增多,並在光鏡下經PAS染色即可發現。另在電鏡下還可發現異常的線粒體,甚至顯示晶格狀包涵體。
另外,應注意正常人類肌細胞中可含稀疏微量的脂滴形態測量學研究發現其含量少於細胞容積的0.2%,故較易區分。
臨床表現
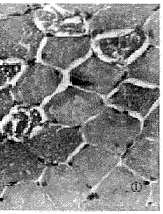 頸肌
頸肌如為肉鹼缺乏致病,且屬全身性者則除表現進行性四肢近端骨骼肌無力外同時有心肌病,並常伴低酮性低血糖等全身性徵候。
因肉鹼棕櫚醯基轉移酶缺乏引起的LSM以Ⅰ型CPT缺乏者最為常見屬常染色體隱性遺傳,基因定位於1ql2多在青少年時期發病,男性發病率較女性高。Ⅰ型CPT缺乏者臨床特點為肌痛肌無力、肌痙攣持久運動和長時間空腹可引起肌肉發硬及發作性肌紅蛋白尿。約1/4的患者導致腎功衰竭一般女性患者症狀較輕。
檢查
 血清肌酸磷酸酶
血清肌酸磷酸酶血清肌酸磷酸激酶(CK)多顯著升高。其他肌酶如乳酸脫氫酶等也多顯著升高Ⅰ型CPT缺乏的患者在肌紅蛋白尿發作時,血清CK可同時升高。
其它輔助檢查:
肌電圖檢查多呈肌源性損害。
治療
1.口服潑尼松治療成人開始劑量為20~40mg/d晨間一次服下,1個月後隨病情改善而漸減劑量減量速度宜慢並隨劑量減少而服用時間延長以免因減量過快導致病情反覆維持量為5~10mg/d,可連用數月兒童劑量酌減。潑尼松療效的機制尚不明確可能對三醯甘油脂肪酶(triglyceridelipase)有直接激活作用(Engel,1972)或由於潑尼松可刺激肌細胞對肉鹼的攝取(Molstad等,1979)。
2.對肯定為肉鹼缺乏者可口服L-肉鹼作為替代療法。開始劑量為100mg/kg以後減為25mg/kg每4~6小時1次。對肉鹼棕櫚醯基轉移酶缺乏所致的患者尚無特殊療法。
3.如有肌紅蛋白尿及腎功能衰竭應採取對症治療宜進食低脂、高糖飲食,並應避免持久運動及空腹飢餓。
病例
 股骨頭
股骨頭查體:血壓19/13.3kPa,脈搏92次/分,體溫36℃,呼吸20次/分。發育正常,營養差,心肺聽診未見明顯異常。神智清,語音低微,雙眼球向各方向活動自如,雙咀嚼肌力弱,雙鼻唇溝對稱,軟齶上舉對稱,咽反射尚存在。雙側轉頭聳肩無力。四肢全般性消瘦,近端肌力3級,肌張力低,遠端肌力4級,無肌束顫動,無肌肉握痛及假性肥大。四肢腱反射消失,病理反射未引出,無感覺障礙。
實驗室檢查:CPK470.3IU/L,LDH259.0IU/L。肌電圖未見特徵性改變,重複頻率刺激無明顯遞增或遞減。心電圖為左前束支傳導阻滯。肝B超為脂肪肝。右肱二頭肌活檢,恆冷切片,經HE、MGT、油紅O、PAS染色後光鏡觀察,示肌纖維大小不等,萎縮的多為Ⅰ型纖維,其內可見空泡或裂紋狀。油紅O染色Ⅰ型纖維內見大小不等的脂滴。MGT未見破碎紅纖維,PAS亦未見特徵性改變。
討 論 脂質沉積病是肌肉中長鏈脂肪酸代謝障礙引起脂質在肌纖維內沉積的一組肌病,與常染色體隱性遺傳有關。該病自1972年由Engel等詳細報導後,才被人們真正認識。多數學者認為,脂質沉積病是脂質代謝性疾病。主要由於肉毒鹼或肉毒鹼棕櫚醯基轉移酶缺乏,導致肌細胞中自由脂肪酸不能進入線粒體進行能量代謝,從而沉積在肌細胞所致。導致肌纖維內脂肪沉積的原因是脂肪代謝的不同環節發生異常。感染、分娩以及某些藥物可以誘發此病。肌肉活檢脂肪染色是診斷該病的主要方法之一。本病例有四肢近端肌無力,有咬肌及頸項肌無力,肌肉病理證實肌纖維內脂質沉積,符合肉毒鹼缺乏症Ⅰ型的診斷。該患病史長達15年,近3年才得到正確治療,提示臨床醫生對疑及本病病人應早期肌肉活檢確診。激素治療會使病人症狀得到明顯改善。治療上應注意的問題是應首先除外可因套用激素加重的疾病,如糖尿病、消化道潰瘍及結核病等。應進食富含肉毒鹼的牛羊肉和牛奶製品,改一日三餐為一日多餐,以減少糖原轉化為脂肪。低脂肪高碳水化合物或含中鏈及短鏈脂肪酸飲食,可減少脂肪在肌纖維內過多的沉積。
