分子結構
維生素B12分子的核心是一個與鈷離子配位的咕啉環結構(圖中用紅色標出)。與該家族的維生素中有含鈷配體和含氰配體維生素有關的全合成稱作氰鈷胺全合成。咕啉環的邊緣通過C1和C2間隔基與甲基(8)和醯胺基(9)相連。第七個醯胺基是長鏈的N-烷基,其中包含一個異丙醇基、一個磷酸基、一個核糖基和一個二甲基苯並咪唑基。咪唑環上的一個氮原子是與中心鈷原子配位的第五個氮原子。咕啉環上總共有多達九個手性碳原子,這給它的合成增加了難度。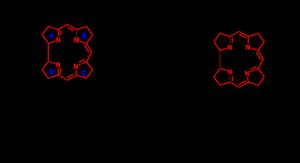
逆合成分析
逆合成的第一步很簡單。早在1960年,伯恩豪爾就提出尾狀的長鏈可以用醯胺水解反應從維生素B12中離去,得到鈷啉胺酸後可以再替換上其他基團。伍德沃德與艾申莫瑟嘗試的是一項嚴格的中繼合成,因為合成目標只是鈷啉胺酸,並沒有包括前人已經完成的尾狀長鏈的連線。
C5和C15上的甲基只有在咕啉環核心構建完成才能引入。該核心可由左側的AD部分(III)和右側的BC部分(IV)合成。由於空間位阻,兩部分直接成環是不可行的,而在含硫試劑作用則可行。
合成路線
A環合成
A環的合成首先由間甲氧基苯胺(1)和乙偶姻(2)進行縮合反應得到希夫鹼甲氧基甲基吲哚(3),然後與炔丙基碘(4)的格氏試劑反應生成炔丙基假吲哚(5)。該物質在三氟化硼和氧化汞的催化下於甲醇中反應,經過甲基被迫處於順式的中間體(6)(親電加成)發生關環反應得到(7)。該化合物以兩種對映異構體的外消鏇體形式存在,使用(-)-α-苯乙基異氰酸酯進行手性拆分可以分離出(+)鏇光性的異構體。
D環合成
D環的合成是從手性的(S)-樟腦(8)在羥胺作用下轉化為肟(9)開始的,然後通過水解轉化為醯胺(10),繼續反應得到內醯胺(11)(酸-胺縮合),N-亞硝基化合物(12)、重氮化合物(13)和環戊烯衍生物(14)(卡賓甲基插入)。使用氫化鋁鋰還原得到醇(15),再用鉻酸氧化得到醛(16),然後用(亞甲基甲酸甲酯)三苯基膦進行一次Wittig反應生成反式烯烴(17),最後水解生成羧酸(18)。AD環偶聯
胺(7)和羧酸(18)在醯氯作用下縮合成醯胺(19),然後在叔丁醇中用叔丁醇鉀處理,發生Michael加成反應得到氫原子處於反式的三環化合物(20)。對將要進行芳香環部分還原的底物上保護基,其中一個羰基與乙二醇反應生成縮酮 (21)另一個與亞胺離子的鹽(氟硼酸三乙基覡)在甲醇鈉的甲醇溶液中反應生成烯醇醚(22)和原醯胺(23)。在甲苯中加熱逸出甲醇可得到烯醇醚(24)。Birch還原反應生成四烯(25),再用酸處理得到二酮(26),也被稱為異環戊烯酮(pentacyclenone)。26中的第二個保護基乙縮醛通過酸解轉換為酮(27)。單肟(28)(位阻比酮羰基更大)可以由二肟的選擇性水解(亞硝酸或乙酸催化)製得。新的第二個氮原子是構建AD環關環時需要的。環戊烯環以及環己烯酮環經臭氧化反應氧化生成三酮(29),然後1,5-二酮單元進行一次羥醛縮合反應(四氫吡咯乙酸酯)生成六元環(30),並伴隨著肟基的甲苯磺醯化,用高碘酸再次氧化打開六元環,最後用重氮甲烷酯化得到酯基(31)。通過Beckmann重排(甲醇、聚苯乙烯磺酸鈉、2小時、170 °C)得到未拆分的內醯胺(32),它進一步經胺羰縮合反應、羥醛縮合反應得到四環化合物(33)α-咕啉去甲基甾酮(α-corrnorsterone)(中文名稱的來歷詳見注釋)。這種內醯胺化合物不易開環,因為丙酸酯側鏈的立體化學不相符。α化合物因此在過量的鹼作用下被轉化為它的差向異構體(34),然後再次酸化並用重氮甲烷處理。差向異構體隨後同時與甲醇和苯硫酚反應轉化成35。這確保了咪唑尾狀長鏈的特異性。然後進行臭氧化反應生成醛(36),同時氨將硫酯轉化為醯胺,再用硼氫化鈉進行醛的還原反應、甲磺醯化,然後用溴化鋰溴化得到溴化物(37)。最後一步將醯胺基脫水成腈基完成AD環合成。
C環合成
C環合成的起始原料是手性(+)-樟腦醌(38),然後三氟化硼在乙酸酐存在時與之加成,使其轉化為三甲基環己烯酸乙酯(39),這個反應最早由瑪拿西(Manasse)和塞繆爾(Samuel)於1902年首創。下一步酯水解成羧酸(40),然後醯胺化成為醯胺(41),再進行臭氧化反應得到有機臭氧化物(42)。該物質可用鋅和甲醇還原成丁二醯亞胺(43),然後用鹽酸的甲醇溶液處理生成內醯胺(44),最後熱解完成C環的合成(45)。B環合成
B環合成的起始原料是3-甲基-4-氧代-2-戊烯酸(46),它與丁二烯在氯化錫作用下經Diels-Alder反應形成外消鏇的六元環化合物(47)。該反應是立體專一性的,甲基和羧基處於順式,此後這被證明是一個對鏇電環化反應。使用α-苯乙胺進行手性拆分得到有光學活性的異構體(47)。用鉻酸氧化雙鍵生成三元羧酸中間體(48),經兩次分子內酯化得到雙內酯(49)。通過一次Arndt–Eistert反應在羧酸α位插入一個亞甲基(50),再與氨反應生成內醯胺(51),最後與五硫化二磷反應獲得硫內醯胺(52)。BC環偶聯
B環(52)和C環(45)在過氧化苯甲醯和鹽酸作用下形成硫橋鍵(53)。先進行一次Eschenmoser去硫反應硫原子在亞磷酸三乙酯的位阻影響下形成烯胺和亞胺(54),然後內醯胺基團在氟硼酸三乙基覡和硫化氫存在下轉化為硫內醯胺(55)。AD、BC環偶聯
圖中右側的BC分子(氰溴化物(37))和左側的AD部分(丙酸酯部分外消鏇化的硫代右前體(55)(thiodextrolin,名稱的來歷詳見注釋))在叔丁醇鉀的催化下,經過一個硫離子中間體生成硫醚(56)。然後使用氰乙基膦、三氟乙酸和環丁碸進行合成過程的第二次Eschenmoser去硫反應,得到氰基咕啉內酯前體(57)(cyanocorrigenolide),同時C環的丙酸酯基團也被外消鏇化。由於兩部分巨大的空間位阻,這樣的偶聯是至今為止唯一成功的方法。內醯胺和內酯基團在五硫化二磷、4-甲基吡啶和氟硼酸三乙基覡作用下生成二硫代氰基咕啉內酯前體(58)(dithiocyanocorrigenolide)和S-甲基衍生物(59)。然後二甲胺與之加成並打開硫內酯環,並通過硫離子從甲基上的消除生成一個環外烯鍵。一個早期模板定向合成該化合物的例子以鈷加合物的形式分離出它。最後由(60)到(61)的成環反應是受形成鈷配合物推動的,這是在二氮雜二環壬烷和二甲基甲醯胺的條件下進行另一種的去硫反應。該反應伴隨著環C中丙酸酯基團的外消鏇化。再用碘和乙酸氧化形成內酯(62),並恢復B環的尾狀丙酸酯基團的正確立體化學性質。
最後的努力目標是在5號位和15號位引入甲基。因為10號位被充分禁止,該物質與苄基氯甲基醚反應生成雙(氯甲基)加合物,後者可用苯硫酚進一步轉換成二硫代苯基化合物(63),產物分離需要使用薄層色譜法。然後用雷尼鎳加氫脫硫,該還原反應同時將內酯環打開使之成為羧酸,再用重氮甲烷與之反應生成酯(64)。

在這個階段,混合物異構體的數目通過高效液相色譜法減少到兩個,也就是(65)中C環13號位的外消鏇丙酸酯基團。與硫酸的反應將氰基轉化為醯胺基(66),但C環13號位的立體化學性質再次被破壞。產量較少的所需異構體(67)再次用高效液相色譜法分離出來。醯胺基然後被來源於氯乙醛的環己基硝酮(cyclohexylnitrone)和四氟硼酸銀轉化為羧基(68)。最後一步中六個酯基與氨合氯化銨反應轉化成鈷啉胺酸中的醯胺基(69)。

