疾病簡述
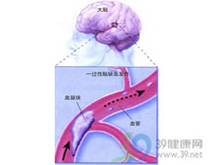 皮克病
皮克病痴呆的發病率和患病率隨年齡增長而增加國外調查顯示,痴呆的患病率在85歲以上人群中高達40%以上其中阿爾茨海默病(Alzheimer’sdiseaseAD)占50%;多灶梗死性痴呆(也稱血管性痴呆,VaD)占12%~20%其餘類型痴呆占15%~20%我國60歲以上人群痴呆患病率為0.75%~4.69%伴隨人口的老齡化,痴呆的絕對及相對發病率明顯增加,皮克病也屢見病例報告但尚未見分類性發病率統計資料。
發病機制
 皮克病
皮克病大體病理觀察腦重量減輕女性平均1050g男性1075g腦萎縮累及額、顳葉(占54%)或單獨侵犯額葉(25%)或顳葉(21%),約1/3的患者雙側對稱受累,約1/2左半球嚴重受累1/5以右半球受累為主顳上回後2/3常不受累是皮克病的特徵之一杏仁核受累較海馬明顯黑質及基底核可受累灰質和白質均可累及,側腦室前角、顳角輕中度擴大光鏡下觀察萎縮的腦葉皮質各層神經元數目明顯減少Ⅱ、Ⅲ層顯著,ⅤⅥ層較輕;部分病人可見頸髓及胸髓運動神經元丟失皮質及皮質下白質星形膠質細胞瀰漫性增生伴海綿狀改變,殘存神經元不同程度的變性萎縮,部分神經元胞質內含界限清楚的嗜銀Pick小體部分神經元表現為膨脹變性的Pick細胞電鏡下Pick小體界限清楚,呈圓形或卵圓形無包膜,是直徑5~15μm的嗜銀包涵體由10nm細絲核糖體、囊泡、脂褐素及24nm短節段直的及扭曲的神經微絲及微管組成,Pick小體的形成機制不清,高爾基浸染法可見皮質錐體細胞樹突棘幾乎完全消失。
臨床表現
 皮克病
皮克病2、皮克病的臨床經過可分為3期(表1)早期以明顯人格改變情感變化和行為異常為特徵,表現為易激惹暴怒固執情感淡漠和抑鬱情緒等漸出現行為異常、性格改變舉止不適當缺乏進取心對事物漠不關心及衝動行為等;部分患者出現Klüver-Bucy綜合徵表現為遲鈍、淡漠順從、視覺失認口部過度活動(hyperorality)、思維變化過速(hypermetamorphosis)善飢和過度飲食把撿到的任何東西如廢紙垃圾和糞便等都放入口中試探和吃掉並有情感抑鬱、焦慮軀體異常感和片段妄想等。
3、隨病情進展可出現認知障礙,逐漸不能思考注意力和記憶力減退與Alzheimer病相比認知障礙不典型空間定向保存較好記憶障礙較輕言語能力障礙較明顯,逐漸言語減少、辭彙貧乏刻板語言、模仿語言和失語症,後期可出現緘默症。
4、神經系統體徵如吸吮反射、強握反射可在病程早期出現晚期發生肌陣攣錐體束及錐體外系損害如帕金森病綜合徵等。
鑑別診斷
 皮克病
皮克病1、中老年人(通常50~60歲)早期緩慢出現人格改變、情感變化和舉止不當逐漸出現行為異常,如Klüver-Bucy綜合徵。
2、言語障礙早期出現,如言語減少、辭彙貧乏、刻板語言和模仿語言隨後出現明顯失語症,早期計算力保存記憶力障礙較輕,視空間定向力相對保留。
4、CT和MRI顯示額和(或)顳葉不對稱性萎縮。
5、病理檢查發現Pick小體和Pick細胞。
 皮克病
皮克病具備1~4項排除其他痴呆疾病臨床可診斷為額顳痴呆如有家族史遺傳學檢查發現tau蛋白基因突變可確診;具備1~5項可確診為皮克病。
皮克病主要應與Alzheimer病鑑別二者均發病隱襲進展緩慢臨床上有許多共同點。最具有鑑別意義的是進行性痴呆症狀在病程中出現的時間順序AD早期出現遺忘、視空間定向力和計算力受損等認知障礙社交能力和個人禮節相對保留;皮克病早期表現為人格改變言語障礙和行為障礙,空間定向力和記憶力保存較好,晚期才出現智慧型衰退和遺忘等。Klüver-Bucy綜合徵是皮克病早期行為改變的表現AD僅見於晚期CTMRI有助於兩者的鑑別,AD可見廣泛性腦萎縮皮克病顯示額和(或)顳葉萎縮;臨床確診需組織病理學檢查。
實驗檢查
測定腦脊液血清中ApoE多態性、Tau蛋白定量β澱粉樣蛋白片段,有診斷或鑑別診斷意義。
1、腦電圖檢查早期多為正常,少數可見波幅降低,α波減少;晚期背景活動低α波極少或無可有不規則中波幅δ波,少數病人有尖波,睡眠時紡錘波少很少出現κ綜合波慢波減少。
2、CT和MRI檢查可見特徵性局限性額葉和(或)顳葉萎縮腦回窄腦溝寬及額角呈氣球樣擴大,額極和前顳極皮質變薄,顳角擴大,側裂池增寬,多不對稱少數可對稱,疾病早期即可出現SPECT檢查呈不對稱性額、顳葉血流減少PET顯示不對稱性額顳葉代謝降低,二者較MRI更敏感,有助於早期診斷。目前尚無有效療法主要是對症治療乙醯膽鹼酯酶抑制劑通常無效。對有攻擊行為易激惹和好鬥等行為障礙者,可謹慎地使用小量苯二氮類、選擇性5-HT再攝取抑制劑精神安定劑和普萘洛爾(心得安)等有條件者可住院治療,或由經培訓的照料者給予適當的生活、行為指導及對症處理。
相關疾病
 皮克病
皮克病A型和B型(也稱鞘磷脂沉積症)以常染色體隱性遺傳為特徵,多出現在猶太人家庭中,A型的特徵是肝,脾腫大,生長障礙,快速的進行性的神經元變性,這常導致2~3歲前的死亡.A型病人(神經)鞘磷脂的活性常低於5%,B型比A型有更多的臨床表型.臨床上常有黃色瘤,皮膚色素沉著,肝脾腫大及淋巴結病。常存在血細胞減少,很多病人常無或有很輕的神經系統受累而能夠存活到成人期,嚴重的B型病人,進行性的肺部浸潤為最主要的併發症。
C型為常染色體隱性遺傳,發生於整個家族中,臨床表現包括肝,脾腫大,進行性的共濟失調,癲癇發作,(肌)張力障礙,有時會出現致命的新生兒肝臟疾患,常在兒童期的晚期出現症狀,10~20歲死亡。成年人有時出現精神病和痴呆。
B型病人常在兒童期因肝脾腫大而得到診斷,A型和B型可以通過組織活檢測定(神經)鞘磷脂酶的活性而確診,標本活檢和組織培養可以證明(神經)鞘磷脂的缺陷.血脂常正常.對A型和B型病人可以通過羊膜或絨毛膜穿刺進行產前診斷。C型診斷需要檢測細胞的膽固醇酯化和在成纖維細胞培養中,暴露於LDL的菲里平膽固醇染色特徵.三型的治療除支持治療外無特殊治療。
皮克病(Pick'sdisease)和阿爾茨海默病(Alzheimer'sdisease,AD)同屬老年性痴呆。近來在AD研究中認為β-澱粉樣蛋白(β-AP)的沉積引起神經細胞變性、缺失,是AD發病的中心環節[1,2]。有人發現在其他神經變性疾病病人腦中β-AP沉積亦有增加。對4例皮克病病人的額、顳葉腦組織切片用ABC法檢測β-澱粉樣蛋白4(β-Amyloidprotein4,βA4)在腦內的表達,旨在探討βA4在皮克病病人腦中表達水平、分布及與變性疾病的關係。
舉例說明
 皮克病
皮克病研究對象
屍檢腦組織標本,經病理診斷證實為皮克病。
實驗方法
①免疫組織化學染色:將上述病例顳葉(21區)和額葉(11區)腦組織標本石蠟包埋,切片4μm,用ABC法行免疫組化染色。依次加入第一抗體(鼠抗人βA4單克隆抗體),第二抗體,AB複合物,DAB顯色。實驗主要步驟:在37℃水浴條件下,一抗、二抗、AB複合物分別孵育切片120min、60min、30min,每一步驟間均以0.01MpH7.4PBS液沖洗,DAB顯色5~10min,並鏡下控制。每例同時設立陽性對照,陰性對照。②對以上標本同部位切片作剛果紅,LHE染色。剛果紅染色設陽性對照。③βA4活性判定方法:在100倍鏡下隨機選5個視野行βA4陽性斑塊記數,求其平均值。依βA4免疫組化染色陽性斑塊數分4個等級:“-”表示陰性,“+”表示弱陽性,每視野平均斑塊數為1~30,;“+ +”表示陽性,每視野平均斑塊數為31~60;“+ + +”表示強陽性,每視野平均斑塊數為61個以上。
2、結果
 皮克病
皮克病從表1可看出βA4主要在皮質第3、5層,在血管、毛細血管及神經元內表達,而白質基本無βA4的沉積。其分布類似AD:①神經元內:βA4陽性斑呈淡棕色,主要在皮質3、5層神經元內表達。②血管:βA4陽性的血管主要分布在大腦皮質第3、5層,斑塊有些顯棕黑色,覆蓋血管;有些呈淡棕色。3例病人血管上βA4表達為陽性。③血管周圍:斑塊呈淡棕色,沙樣型。只有1例在血管周圍表達陽性。④毛細血管:2例βA4有陽性斑塊的毛細血管主要分布在皮質3、5層,有些呈棕黑色、有些呈淡棕色。⑤毛細血管周圍:βA4陽性斑呈淡棕色,沙樣分布。1例呈現上述特徵。剛果紅染色:所有病例均為陰性。
βA4的病理類型
病理類型依βA4陽性斑的形態將其分3型,Ⅰ型為典型大核心型。斑塊為深棕色,中間有一大的深棕色核,核心象一個死亡的神經元。主要分布在皮質第3層。Ⅱ型為典型小核心型。斑塊呈棕黑色,有一小圓形的棕黑色核心,中心貌似有一血管。主要分布在皮質第5層。Ⅲ型為非典型沙樣型。斑塊為淡棕色,沙樣分布,此型斑塊好象為棕色物質從死亡的神經元、血管內漏出。主要分布在皮層第3、5層,尤其在神經元中。皮克病主要以典型大核心型和非典型沙樣型為主。
