流行病學
本病重型多在2~3歲發病,輕型病例發病年齡較大。目前沒有其他相關內容描述。病因
本病常染色體隱性遺傳。發病機制
本病的基本生化缺陷是N-乙醯半乳糖胺-4-硫酸酯酶活性減低,它可把N-乙醯半乳糖胺-硫酸醋中的硫酸水解掉此反應既發生在軟骨素-4-硫酸,也發生在硫酸皮膚素該基因定位於5q12~q13,可能有2個等位基因本型人體內由於這種酶的活性不足,故可使軟骨素-4-硫酸和硫酸皮膚素沉積於各種組織細胞內而致病。臨床表現
 黏多糖貯積症Ⅵ型
黏多糖貯積症Ⅵ型根據臨床症狀的輕重程度,可將這型黏多糖貯積症分為重型(MPSⅥ-A型)和輕型(MPSⅥ-B型)。重型多在2~3歲發病,常有關節運動受限、疝氣、四肢攣縮、容貌粗笨、短頸、身材矮小。因軀幹和四肢發育遲緩故可出現身材比例失調容貌粗笨不及黏多糖貯積症Ⅰ型和Ⅱ型那樣明顯頭顱略大,兩眼間距過寬。部分病例也可合併腦積水。大多數病例可有不同程度的周邊性角膜渾濁和聽力消失。也可出現鍾狀胸、漏斗胸、臍疝和腹股溝疝。在主動脈瓣區常可聽到主動脈關閉不全而產生的雜音。一般有肝、脾輕度到中度腫大可引起脊柱、骨盆手和頭顱等全身骨骼改變,第2頸椎發育不良,可導致寰樞脫位和腦膜增厚,也可發生脊髓病或神經系統併發症,胸腰部椎體變形骨盆發育不良。本型病人同其他幾型黏多糖貯積症一樣也可出現腕骨發育不全、變形及掌骨呈棒狀改變人字縫融合和齒狀突發育不良可引起神經併發症,如腦積水和痙攣性截癱。由於膠原組織壓迫神經,常出現腕管綜合徵。可反覆發生呼吸道感染。有的可發生心力衰竭。輕型病例發病年齡較大骨骼畸形較輕病情發展亦較緩慢。
併發症:
本病的重型可並發腦積水和痙攣性截癱,有的可發生心力衰竭,臍疝和腹股溝疝肝脾輕度到中度腫大。
診斷
本型的診斷依據是出現典型的臨床症狀,尿中排出過多硫酸皮膚素和白細胞出現明顯的異染性顆粒(Reilly小體)白細胞或皮膚或纖維細胞培養顯示N-乙醯半乳糖胺-4-硫酸酯酶缺乏。
鑑別診斷:
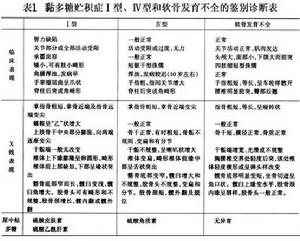
本型的鑑別診斷與Ⅰ型相同見表1。
檢查
實驗室檢查:
尿液中有過多的硫酸皮膚素。中性粒細胞和淋巴細胞有多而明顯的異染性顆粒。
其它輔助檢查:
X線檢查:同黏多糖貯積症Ⅰ型以四肢、脊柱、骨盆改變為常見。此外尚有頭顱改變,並具有特徵性。
1.四肢 股骨和脛骨管狀骨有骨幹異常,許多病例有骨骺板和千骺端畸形。上肢長管狀骨較下肢改變明顯手和足的短骨改變因人而異,但均有畸形,表現掌骨呈棒狀,腕骨發育不全或呈畸形。
2.脊柱 以胸腰椎處改變為明顯,表現有椎骨小,呈鳥嘴狀或為其他畸形。也可有脊柱後凸。肋骨呈船槳狀,伴有胸骨向前突出可使胸部呈漏斗狀肋骨變形可使胸部呈鍾樣。
3.骨盆 髂骨翼發育不全,並有畸形髖臼小股骨頭骨骺不規則和變扁。
4.顱骨 蝶鞍增大呈“乙”形有硬化性乳突、齒狀突發育不全及人字縫融合。
治療
同黏多糖貯積症Ⅰ型。對關節攣縮、胸廓畸形,可行手術矯形治療,對易感染傾向也必須進行有效的防治。至於其他症狀如心力衰竭等,可以進行對症治療。
預後:
因人而異,重型病人一般在10歲前死亡,輕型病人則可長期存活。
預防:
糖原累積病是一組兒童遺傳性糖原代謝紊亂的疾病,主要以機體組織糖原累積過多而分解困難為特點,極少為糖原合成代謝障礙,致使機體組織糖原儲存少。糖原累積病並非為一種疾病,而是一組疾病。目前確定的已有12種。臨床均以低血糖為特徵,所涉及到的器官主要為肝、腎、骨骼肌。遺傳方式多為常染色體隱性遺傳,無性別差異,多在兒童期發病。部分病人至成人後,病情不再發展可以維持一般健康水平
患者主要是由於缺乏分解糖原的某些酶,如葡萄糖-6-磷酸酶、α-1,4葡萄糖甙酶、磷酸果糖激酶、肝磷酸化激酶等。
許多患者的父母為近親結婚,避免近親結婚是預防本病的重要環節。一旦發現糖原累積症以防治低血糖為主,膳食少量多餐限制脂肪和總熱量,限制體力活動。血清乳酸高者,宜服碳酸氫鈉防治酸中毒。皮質激素腎上腺素、胰高血糖素等可幫助控制低血糖。
