概述
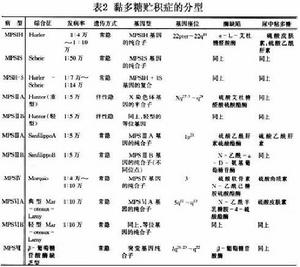
自Hunter(1917)和Hurler(1919)分別報導MPSⅡ型和MPSⅠ型病例以來,人們對本病的認識也不斷深入早期曾稱為‘承霤病’,1952年以後由於在病人肝內發現硫酸皮膚素沉積,並將這類綜合徵改稱為黏多糖貯積症。1957年發現病人尿中黏多糖增多McKusick等(1965)根據病人尿中排出黏多糖的類型不同而將本病分為6型。近年來隨著生物化學和遺傳學的迅速發展證明,黏多糖在細胞內沉積是發生在溶酶體內。人們掌握了培養成纖維細胞的技術之後,對於了解MPS的生化異常和發現不同的遺傳類型都有重要影響。現已闡明MPS的7個類型12個病種的臨床和生化特點。每一個類型都由不同的基因突變所引起。現將本病分類列表介紹如下(表1)。
黏多糖貯積症Ⅰ型本型又稱胡勒(Hurler)綜合徵、承霤病、MPSI-H。既往曾稱脂肪軟骨營養不良、軟骨-骨營養不良。胡勒(Hurler)於1917年首先描述,國內自1955年以後也有報導。
流行病學
黏多糖貯積症Ⅰ型多發病於幼年,嬰兒、兒童期多見。目前沒有其他相關內容描述。
病因
本病病因是常染色體隱性遺傳。
發病機制
 黏多糖貯積症Ⅰ型
黏多糖貯積症Ⅰ型本病發病機制為常染色體隱性遺傳。其基本缺陷是溶酶體a-L-艾杜糖醛酸酶缺乏基因定位在22~22q11,從而使黏多糖分解發生障礙體內各種組織細胞內有分解不全的黏多糖沉積,並隨尿排出體外。從尿中排出的2種黏多糖是硫酸皮膚素和硫酸乙醯肝素,二者在多糖側鏈上都有艾杜糖醛酸。
病理:本型的病理改變幾乎可涉及到體內的每個器官和組織,其中最嚴重的改變見於腦、心、肝脾。腦體積增大,重量增加,軟腦膜變厚,呈乳樣渾濁。心臟肥大,心包心內膜、腱索、瓣膜都有片狀或結節狀肥厚。冠狀動脈呈白色索狀可被內膜沉積所阻塞,或呈管腔狹窄肝脾腫大變硬,外觀呈灰色。
鏡下可見在許多組織內都有腫脹的大細胞內有大量黏多糖沉積,稱為Hurler細胞或氣球樣細胞。這種細胞可能屬於成纖維細胞巨噬細胞或其他細胞見於心瓣膜、血管、腦膜角膜、肌腱和骨膜等組織由於黏多糖溶解於福馬林,故用PAS染色時,黏多糖沉積溶解。肝實質細胞、庫普弗細胞脾和淋巴結的網狀細胞、內分泌腺的上皮細胞、軟骨細胞、骨細胞和視網膜細胞等,也有黏多糖沉積,可使細胞呈空泡狀,與Hurler細胞相似。用組織化學方法分析細胞沉積物證明是黏多糖。電鏡研究提示異常細胞內沉積物是在溶酶體內。
中樞神經和末梢神經系統的神經元和膠質細胞腫脹,內有脂樣沉積物,其組織化學特點與神經節苷脂相同。電鏡檢查可見神經元內沉積物常聚結或為分層結構,稱斑馬小體,與神經節苷脂病時的膜狀胞漿小體很相近。
臨床表現
 黏多糖貯積症Ⅰ型
黏多糖貯積症Ⅰ型本症在內臟病變、骨骼畸形和智力障礙方面的症狀都很嚴重。患兒初生時外表尚正常但以後可很快表現出運動和智力發育落後至1~2歲時可明顯顯露出本病的特徵。患兒容貌逐漸變得粗笨,鼻樑寬而平,眼距寬,唇厚,舌大,耳低位牙齒小而稀疏齒齦肥厚。皮膚粗厚毛髮增多,眉睫毛長。頭圍增大矢狀縫早閉前後徑增大(舟狀頭),偶有腦積水骨關節嚴重畸形,手指粗短短頸,脊柱後彎。各關節逐漸攣縮強直,手指固定於半屈位,呈爪狀身材矮小。
1.神經系統 主要是智力低下,1歲以後即很明顯,行動拙笨,語言發育落後對周圍環境反應遲鈍。智力低下的程度與腦脊液中黏多糖的濃度有關。有驚厥發作者不多。其他神經系體徵,如腱反射減低或亢進、痙攣性癱瘓病理反射等也都可能出現
2.循環系統 心血管症狀很明顯,可有心臟肥大、肺動脈高壓以及由於瓣膜病變而引起的胸骨左緣或心尖部收縮期雜音。冠狀動脈廣泛梗死,可引起猝死。
3.呼吸系統 反覆發生呼吸道感染,並有通氣障礙。鼻咽分泌物增多,扁桃體和腺樣體增殖顱面骨畸形,可致鼻咽腔狹窄、呼吸困難張口呼吸、有鼾音。若胸廓畸形伴有支氣管軟骨畸形,則呼吸困難更重,並易致肺炎與支氣管炎。
4.消化系統 多數病人都有腹部膨隆,肝脾腫大、光滑且硬,是本病的一個重要特徵。有時可有臍疝或股疝。
5.五官 視力和聽力也有異常。角膜由霧狀而變為嚴重渾濁,以後可發展至失明。有的有大角膜、眼球突出或視網膜病變。傳導性耳聾可能是由於聽骨畸形所致。
併發症:
1.神經系統可並發痙攣性癱瘓。
2.循環系統 可並發心臟肥大、肺動脈高壓冠狀動脈廣泛梗死,可引起猝死。
3.呼吸系統可並發通氣障礙,肺炎與支氣管炎
4.消化系統可並發肝脾腫大、有時可有臍疝或股疝。
5五官可並發,眼球突出或視網膜病變,傳導性耳聾可能是由於聽骨畸形所致。
診斷
根據本型特殊的臨床表現,結合家庭發病史、病理改變及有關實驗室檢查即可確診。
鑑別診斷:
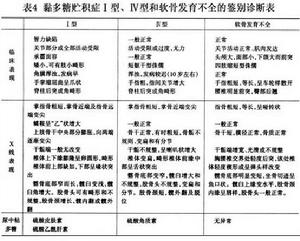
但必要與其他幾型黏多糖貯積症仔細鑑別(表2,3),尤其多糖貯積症Ⅳ型、軟骨發育不全仔細鑑別(表4)。
檢查
 黏多糖貯積症Ⅰ型
黏多糖貯積症Ⅰ型實驗室檢查:
尿中出現過多的硫酸皮膚素和硫酸乙醯肝素。血中白細胞及骨髓細胞內出現異染性黏多糖顆粒(Reilly體)。
其它輔助檢查:
1.X線檢查
(1)四肢骨:由於骨骼內黏多糖過多沉積,成骨和成軟骨活動異常。在管狀骨主要為引起骨幹成型收縮障礙和變短。早期骨幹增粗骨皮質增厚,髓腔狹窄;晚期則皮質變薄髓腔增寬,骨幹的一端或兩端變細,常以生長慢的一端明顯這種改變在上肢較下肢明顯。上肢長管骨粗短中間部分膨隆兩端變細,此為本病特徵性改變之一。肱骨近端呈內翻畸形。由於骨幹兩端呈非對稱性生長,可致骨端彎曲、傾斜,以橈尺骨遠端尤著,其關節面呈相對性傾斜。股骨近端常變細彎曲,可形成明顯的髖內翻畸形,有的可呈髖外翻畸形。股骨遠端受累較輕或反而擴展。手短管骨粗短,掌骨近端收縮、變尖,呈圓錐形。指骨遠端通常變小末節指骨發育不良,骨化不全。掌骨有這些變化時,跖骨也常變為細長。
(2)軀幹:腰1或腰2椎體發育不良、變小及向後移位頗似被擠出。脊柱也以該處為中心向後突出而表現為成角畸形。該椎體前緣上部缺如,下部突出呈喙狀,與黏多糖貯積症Ⅳ型椎體中部呈舌樣突出明顯不同。其他椎體上下面膨隆近似圓形。腰椎體後緣常向內凹陷,椎弓根變細長,骨質疏鬆。髂骨翼向外展開髂骨底部發育不良和變窄,髖臼角增大。肋骨增寬,脊柱端變細,形如划槳。鎖骨內側增寬,外側變細呈“鉤”形。
(3)頭顱:常增大,以腦積水頭型為常見。大多數病例蝶鞍增大,主要為前後徑增大呈“乙”字形,系蝶鞍畸形所致。囟門關閉遲緩。前額及眼眶邊緣突出,兩眼分離過遠。下頜骨發育不全但角度增大。
2.微量酶活性測定 二甲基美藍-Tris(DMB-Tris法)用於尿中糖胺聚糖(GAGs)的檢測及羊水中GAGs的測定,前者多用於患兒的篩查或診斷,後者則用於產前診斷。
3.基因診斷 本病基因座位22~22q11,可用RFLP法或PCR法進行早期診斷。
治療
目前無有效治療方法曾進行過各種試驗治療,但效果不肯定。糖皮質激素在鼠的實驗中有抑制黏多糖合成作用,但病人每天服用潑尼松2mg/kg並未出現臨床效果。大量維生素A加入MPSI-H和Ⅱ型病人培養的成纖維細胞,可以使其異染性消失。給病人口服維生素A 1萬~10萬U,4次/d經1個月~數月後可改善症狀,可使尿中黏多糖排出增多,但與體內沉積的黏多糖量相比則微不足道。維生素C的套用也未見臨床療效。甲狀腺素或生長激素的套用也同樣無效。對於輸入新鮮血漿的治療方法意見尚有分歧,一部分作者認為可得到明顯的暫時的臨床好轉,但其他作者套用卻未得到確切的療效。
預後預防
 黏多糖貯積症Ⅰ型
黏多糖貯積症Ⅰ型預後:
本型為進行性,4歲開始嚴重衰退,常在10歲前死亡死亡年齡較其他類型黏多糖貯積症早,死因多為心功能不全。
預防:
糖原累積病是一組兒童遺傳性糖原代謝紊亂的疾病主要以機體組織糖原累積過多而分解困難為特點,極少為糖原合成代謝障礙,致使機體組織糖原儲存少糖原累積病並非為一種疾病,而是一組疾病。目前確定的已有12種。臨床均以低血糖為特徵,所涉及到的器官主要為肝腎、骨骼肌。遺傳方式多為常染色體隱性遺傳,無性別差異,多在兒童期發病。部分病人至成人後,病情不再發展,可以維持一般健康水平。
患者主要是由於缺乏分解糖原的某些酶,如葡萄糖-6-磷酸酶、α-1,4葡萄糖甙酶、磷酸果糖激酶、肝磷酸化激酶等。
許多患者的父母為近親結婚,避免近親結婚是預防本病的重要環節。一旦發現糖原累積症,以防治低血糖為主,膳食少量多餐,限制脂肪和總熱量限制體力活動。血清乳酸高者,宜服碳酸氫鈉防治酸中毒。皮質激素腎上腺素、胰高血糖素等可幫助控制低血糖。
