流行病學
一般在4~7歲發病,10歲時最嚴重。目前沒有其他相關內容描述。
病因
病因為常染色體隱性遺傳。
發病機制
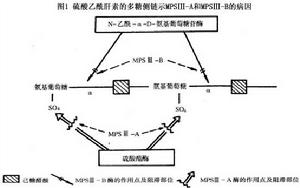 黏多糖貯積症Ⅲ型
黏多糖貯積症Ⅲ型根據成纖維細胞混合培養結果可將本病分為A、B、C、D四型。A型是由於溶酶體乙醯肝素-N-硫酸酯酶缺乏引起;B型是由於N-乙醯-α-D-氨基葡萄糖苷酶(圖1)引起;C型是由於乙醯CoA-α-葡萄糖胺-N-乙醯轉移酶缺乏引起;D型是由於N-乙醯-α-D-氨基葡萄糖苷-6-硫酸酯酶缺乏引起。上述酶缺乏使硫酸乙醯肝素不能逐步降解,而在肝及組織內貯積,並大量隨尿排出。雖然酶的缺陷不同,但4型的症狀完全相同臨床上不能區分。4型細胞可以相互矯正代謝缺陷使4型病人細胞內的黏多糖沉積都得到矯正。這種“交叉矯正”現象大概是因為不同型病人的成纖維細胞可以相互供給細胞缺乏的酶所致。
病理:病理檢查可見肝細胞和庫普弗細胞以及直腸黏膜細胞中有大空泡形成。腦室擴大,腦實質神經元嚴重變性和脫失,而殘留有神經元氣球樣變,內含脂質而外觀腫脹;腦灰質鞘脂大量增多。基底神經節和視神經節細胞變性腫脹。腦的這些病變就是臨床上嚴重智力低下的原因。
電鏡檢查顯示有不同類型的細胞質包涵體;具有均勻顆粒物質及小同心圓性膜小體空泡具有親水外膜和斑馬小體空泡。
組織化學檢查證明腦、肝、腎所沉積的黏多糖主要為硫酸乙醯肝素腦組織中還有糖脂GM神經節苷脂含量增加。
臨床表現
進行性智力低下是本症的最顯著特徵。生後1歲內智力發育均正常或僅有輕度落後,智力低下一般在4~7歲出現,10歲時已很嚴重。在智力低下進行性加重的同時,可出現進行性神經症狀,如抽風運動過多、痙攣性四肢癱、全身無力攻擊性行為等,此為該症最突出的症狀。
身材改變較輕或表現正常,僅有1/4的病人表現矮小。大多數病人面部表現正常,少數病人表現頭大、面容醜陋、腹部膨隆、進行性耳聾、關節僵直和手屈曲呈爪狀。骨骼改變僅有多發性成骨不全和頂骨後部緻密、增厚。這些改變對診斷本病具有一定程度的特異性。肝、脾腫大為輕度到中度,無角膜渾濁。無心臟受累。
併發症:
本病可並發痙攣性四肢癱、攻擊性行為等。即使有肝脾腫大,也很輕微。
診斷
根據臨床特徵和尿液分析結果,應懷疑該型綜合徵的診斷,而確診必須依靠證實有特異的酶缺陷,因為本型4個亞型臨床表現相似。
鑑別診斷:
鑑別診斷的注意事項與黏多糖貯積症Ⅰ型相同。
檢查
實驗室檢查:
尿中排泄的大量黏多糖是硫酸乙醯肝素,MPSⅢ-B病人血清中酶的活性為正常者的2%~16%而雜合子的酶活性在正常者的35%以下。血液中淋巴細胞、中性粒細胞和骨髓細胞可發現有異染性包涵體。
其它輔助檢查:
X線檢查:X線表現與黏多糖貯積症Ⅰ、Ⅱ型相似,約半數病人可出現Ⅰ型改變但程度較輕,其中僅1/4的病人發生侏儒另外半數病人可出現長骨和肋骨增大畸形。部分病人腰椎椎體呈雙凸形,頂骨後部密度增高、增厚。
治療
同黏多糖貯積症Ⅰ型加強出生前診斷為本症的惟一預防措施。
預後預防
預後:
可能在兒童期死亡,也可存活較長,多死於呼吸道感染。
預防:
糖原累積病是一組兒童遺傳性糖原代謝紊亂的疾病主要以機體組織糖原累積過多而分解困難為特點,極少為糖原合成代謝障礙致使機體組織糖原儲存少。糖原累積病並非為一種疾病,而是一組疾病。目前確定的已有12種。臨床均以低血糖為特徵,所涉及到的器官主要為肝、腎、骨骼肌。遺傳方式多為常染色體隱性遺傳,無性別差異,多在兒童期發病。部分病人至成人後病情不再發展,可以維持一般健康水平
患者主要是由於缺乏分解糖原的某些酶,如葡萄糖-6-磷酸酶、α-1,4葡萄糖甙酶、磷酸果糖激酶、肝磷酸化激酶等。
許多患者的父母為近親結婚,避免近親結婚是預防本病的重要環節。一旦發現糖原累積症,以防治低血糖為主,膳食少量多餐限制脂肪和總熱量,限制體力活動。血清乳酸高者,宜服碳酸氫鈉防治酸中毒。皮質激素、腎上腺素、胰高血糖素等可幫助控制低血糖。
