 蛋白粒子病
蛋白粒子病蛋白粒子病(prion disease)是一組可以累及人和動物中樞神經系統的、可以傳播的慢性變性疾病。曾被稱為可傳播的神經系統變性病,亦曾稱為神經系統慢病毒感染。近年研究證明,本組疾病是由一種可傳播的蛋白酶(朊酶)微粒——蛋白粒所造成。Prion是一種蛋白酶,它不含核酸,其基因編碼位於人染色體20的短臂上。正常人細胞的蛋白粒子稱為PrPc,它分為膜相關型和分泌型,在正常神經元中有大量等位形式存在的PrPc,它在發育過程中調控。在不明原因的條件下,PrPc轉換成PrPsc,它對蛋白水解酶有抵抗,並能自發聚集形成蛋白粒子微桿而致病。
病因
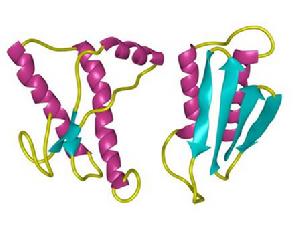 Prion染色體結構,左PrPc,右PrPsc
Prion染色體結構,左PrPc,右PrPsc美國生物學家普魯西內爾因發現引起瘋牛病的朊病毒而獲得1997年諾貝爾醫學獎。是人類正常蛋白分子電腦圖像,會引起瘋牛病的不正常蛋白模型。
1972年,普魯西內爾提出引起動物和人(比較少見)的中樞神經系統變性疾病的傳染性因子可能是蛋白質。人們對此產生懷疑,認為是左道邪說。生物學的教條認為傳染性疾病的傳播因子必須具有DNA和RNA組成的遺傳物質,才能在宿主體內感染。即使最簡單的微生物中病毒也要靠核酸的指令為存活和繁殖所必需的蛋白質的合成。
通過對患瘋牛病牛死後腦解剖病理組織分析結果表明,牛腦灰質中有與羊癢症相關的原纖維存在,感染因子含於其中。普魯西內爾在1982年將感染因子命名為Prion,它是一種蛋白感染顆粒,是一種抗蛋白酶作用的糖蛋白PrP。它具有病毒的特徵,但很多使病毒失活的方法,試劑都對它無效。它與病毒的區別在於它比病毒更小,不含核酸,不會在患病體中誘發產生抗體。這種致病的蛋白粒子PrPsc,是正常蛋白質PrP的突變異構件。
分類
 牛海綿體狀腦病
牛海綿體狀腦病人類Prion病
雅-克氏病(Jacob-Creutzfeldt disease)
Gerstmann-Straussler-Scheinker病(GSS綜合徵)
庫魯(Kuru)病
致命性家族性失眠症
遺傳性伴痴呆性痙攣性截癱
動物Prion病
羊瘙癢症(scrapie)
貂傳遞性腦病
麂和鹿慢性消瘦性肌病
瘋牛病,牛海綿狀腦病(BSE)
雅-克氏病
 CJD
CJD雅-克氏病簡稱CJD,是蛋白粒子病中最常見的臨床類型,曾被稱為皮質-紋狀體-脊髓變性,人腦海綿狀腦病。臨床上有家族遺傳性、散發性、醫源性和新變異型等數種臨床類型,以散發性病例最多,約占85%左右。總體人群中患病率約為百萬分之一至二。
病因與發病機制
CJD是Prion病中最常見的臨床類型。蛋白粒子存在於正常人的神經細胞內,稱為細胞型蛋白粒子(PrPc),該蛋白性質保守,能迅速被蛋白酶K消化。變異型細胞型蛋白粒子(PrPsc)位於細胞外面,它與PrPc功能不同,是引起CJD的致病蛋白,能使正常PrPc變為PrPsc,為此CJD疾病一旦起動就很難停止。
CJD的發病與人類基因易患性有關,認為與纈氨酸12q密碼子的多態性有關。凡是密碼子12q為雜合子者患CJD的機會較低,純合子者患CJD的機會較高。
臨床表現
50歲以後隱襲起病,亦有急性或亞急性起病。主要臨床表現為進行性痴呆、四肢肌張力增高,伴肌陣攣發作和手肌肉萎縮。臨床上依症狀可分為枕葉型、丘腦型和小腦型。病人的最早精神症狀為注意力不集中,思維遲鈍,判斷、記憶能力減退,情緒變化大,好發脾氣且反覆無常,還可伴發各種幻覺。90%的患者可發生肌陣攣發作。驚嚇可誘發或加重肌陣攣發作。約有1/3的患者早期出現小腦症狀或視覺障礙,該症狀有時可掩蓋一些精神症狀。此外,隨病程的發展,2/3的患者可出現錐體外系體徵,如震顫、僵直、動作緩慢、笨拙等,但肌腱反射亢進,半數病人錐體束征陽性。整個病程1-2年,少數為數月或2年以上。
體格檢查可見手及肢體肌肉萎縮,肢體有明顯肌陣攣發作。實驗室檢查可見腦脊液中生化常規正常;14-3-3腦蛋白的免疫酶測定陽性。腦電圖檢查可見典型三相波和周期性尖慢波發放,陽性率高達95%。頭顱CT和MRI檢查可見腦室擴大,大腦皮質萎縮。
診斷與鑑別診斷
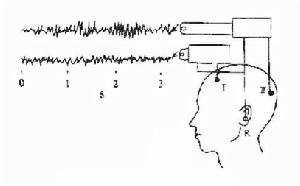 腦電圖三相波
腦電圖三相波根據典型的緩慢起病的進行性痴呆,伴四肢肌張力增高、肌陣攣發作和肌肉萎縮等臨床表現和腦電圖檢查的三相波出現,臨床可以擬診本病之可能。然而,肯定的診斷仍需病理,腦脊液及腦活檢中14-3-3蛋白及PrP蛋白免疫組化染色陽性者可以確診。下列為CJD的診斷標準:
1、臨床表現為:①進行性痴呆;②肌陣攣;③視覺和(或)小腦症狀;④錐體系和錐體外系體徵同時存在;⑤無動性緘默。
2、特徵性腦電圖異常(三相波)。
3、腦脊液中14-3-3蛋白陽性。
具備上述三條者診斷肯定。僅具第一條者可為擬診。
治療
至今無有效治療方法。金剛烷胺或可以延緩疾病進展,但尚無肯定結果。加強護理、減少併發症為本病唯一治療措施。徹底銷毀病人使用的注射器、處理好分泌物是防止醫源性CJD的可靠措施。因此,凡擬診或診斷CJD者均應按傳染性疾病管理辦法予以隔離、銷毀污物處理。
庫魯(Kuru)病
 庫魯氏病
庫魯氏病Kuru病是發生在太平洋島國紐幾內亞東部山區Fore族人群中的可以傳播的神經系統疾病,它與該族有同類相食的宗教儀式有關。也是人類海綿狀腦病成功地傳遞給實驗動物的一種疾病。自1960年禁止同類相食的宗教儀式以來,至1984年以後沒有新病例的發生。
主要臨床表現為嚴重的小腦性共濟失調,伴有自主的多動症,包括舞蹈指划動作、肌陣攣、震顫、構音困難等。後期出現智力障礙,額葉釋放性症狀、摸索、強握等。少數患者伴發痴呆。病人腦組織有明顯萎縮,呈海綿狀變性,腦皮質內有神經元缺失及神經膠質細胞增生。腦組織中可以分離出Prion蛋白,免疫組化染色亦可得到陽性結果。
Gerstmann-Straussler-Scheinker病(GSS綜合徵)
 GSS綜合徵
GSS綜合徵GSS綜合徵是一種十分罕見的遺傳性脊髓小腦變性疾病。1935年由Gerstmann及其同事首次報導。患病率為100萬分之0.02。由PrP基因突變所致,屬常染色體顯性遺傳。常於中年以後,50歲左右起病。
主要臨床表現為運動不穩、行動笨拙、協調困難和進行性痴呆。隨著病程的進展,小腦性共濟失調的症狀加重,並出現構音困難,眼球震顫和錐體外系的帕金森症樣體徵,聽力減退或耳聾;視力減退、甚至失明。實驗室檢查中,腦電圖檢查不出現三相波;但在病理免疫組化中可見PrP蛋白,神經元脫失,星形細胞增生和海綿狀變性。然而,病者生前的臨床特徵很難與橄欖-橋腦-小腦病變相鑑別,亦很難與慢性進展性多發性硬化相鑑別。因此,本病的最終診斷仍有賴於病理學上特徵的免疫組化之結果。本病病程約為3-5年。至今尚無有效治療。
其他蛋白粒子病
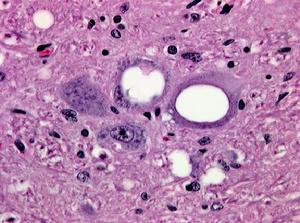 羊瘙癢症的海綿組織損傷
羊瘙癢症的海綿組織損傷其他蛋白粒子病還包括致命性家族性失眠症(FFI),變異型CJD、瘋牛病和瘙癢症等。
FFI
屬於常染色體顯性遺傳,由PrP基因突變引起,突變位置為密碼子178,由Asn代替了Asp而引起突變。本病十分罕見。多數在中年發病。主分臨床症狀為嚴重失眠,伴幻覺和遺忘,自主神經功能紊亂明顯,表現為多汗、心悸、呼吸節律紊亂,血壓升高,體溫升高。病理上主要可見丘腦部神經元脫失、膠質細胞增生。整個病程約在15個月以內,常因衰竭而亡。
變異型CJD
系由1986年在英國出現瘋牛病後提出,它的發病年齡較輕,病程進展較慢,臨床表現的可變性較大,主要特徵為:①精神症狀,表現為焦慮、抑鬱、孤僻、淡漠和行為異常。②共濟失調,常在首發症狀出現後的數周至數個月內出現,表現為小腦性共濟失調。③記憶障礙,常早期出現記憶障礙,以後逐步加重,至晚期痴呆。④肌陣攣。⑤肢體運動感覺障礙,錐體束與錐體外系同時受累的體徵,少數可有輕度肢體或面部感覺障礙。⑥腦電檢查沒有典型的三相波。變異型CJD的病理改變從本質上說與普通CJD沒有區別,但海綿狀變性以基底核最為突出。PrP的免疫組化染色可見大量的Kuru型PrP在灰質區,特別是枕葉、基底核和小腦內大量沉積。因此,變異型CJD的臨床診斷十分困難,最終診斷僅能以病理診斷予以肯定。
瘙癢症
是一種見於綿羊和山羊的一種獨立疾病。1986年英國發現瘋牛病後發現這種在羊中傳播的疾病,可以通過動物飼料傳染給牛,使牛致病。已證實牛亦可傳播給人,因此應當引起注意。
