病因
 神經幹細胞
神經幹細胞進行性肥厚性間質性神經炎為常染色體隱性遺傳,本病多由近親結婚所致。
發病機制:
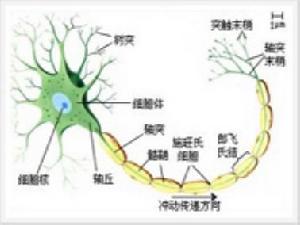
發病機制尚不清楚,主要病理變化為周圍神經乾呈肥厚性改變其形如紡錘,可見結節狀或彌散性變粗,鏡檢可見有髓纖維顯著的節段性髓鞘脫失和再生徵象,施萬細胞呈洋蔥皮樣圍繞,軸索稱之為洋蔥頭樣改變,這種洋蔥頭改變是由於周圍神經受損後,脫髓鞘與髓鞘再生反覆進行的結果。
軸索消失成空心狀m脊髓後索及脊髓前角細胞亦有變性。無髓鞘纖維雖未減少,但可能亦已受累.
流行病學:
尚未查到權威性的較全面的發病率統計學資料本病國內少見,多在嬰兒期起病。可有家族史,亦可為散發性.
症狀
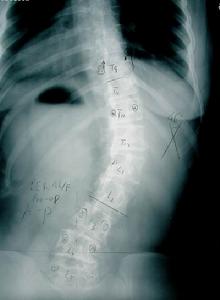 脊柱側彎
脊柱側彎1,本病多在嬰兒期起病可有家族史,亦可為散發性,病程緩慢進展,有時可加重或緩解。
2,初發症狀為下肢無力,輕度肌肉萎縮四肢遠端疼痛感覺異常,痛溫覺減退,位置覺震動覺減退。四肢的神經乾肥大,有壓痛。後期肢體近端肌群亦可受累,表現為無力,肌張力減低腱反射減弱或消失軀幹肌一般不受累及可有肌束震顫。
3,小腦受損亦是本病表現之一,主要為上肢共濟失調如意向性震顫,指鼻不準雙手輪替動作笨拙等;眼球震顫,吟詩樣語言以及閉目難立征陽性等均可出現。其他可有瞳孔縮小瞳孔左右不等大對光反應障礙,弓形足與脊柱側彎等。
4,肌電圖檢查可見運動神經及感覺神經傳導速度明顯減慢。
併發症:
隨病情發展可以出現多樣的症狀體徵。
診斷
 進行性肥厚性間質性神經炎
進行性肥厚性間質性神經炎根據本病在小兒期發病,病程緩慢進展。對稱性感覺、運動神經均受累,以及肢體遠端受累為重周圍神經乾肥大,活檢呈肥厚性間質性神經炎等特點即可確診。
鑑別診斷:
需與下列疾病鑑別:
1,麻風病具有特殊的皮膚損害,查麻風桿菌陽性等可與本病鑑別。
2,腓骨肌萎縮症,本病發病年齡較進行性肥厚性間質性神經炎大,以下肢遠端肌肉萎縮為主,在股中下1/3交界處以下變細,呈“仙鶴腿”,周圍神經一般不肥大,可與本病鑑別。
 進行性肥厚性間質性神經炎
進行性肥厚性間質性神經炎顯性遺傳性HMSNⅡ型可以出現在不同的年齡組,臨床表現和電生理改變在不同病人具有明顯的差異,可分為4個亞型。HMSNⅡA型的致病基因位於染色體1p35~p36位點。發病在老年前期,具有典型的HMSNⅡ型的臨床症狀,表現為進行性的遠端肢體無力和萎縮,症狀一般首先從下肢開始,而後發展到上肢。電生理檢查發現正中神經傳導速度和動作電位波幅下降不明顯,下肢腓腸神經的動作電位波幅出現顯著下降。HMSNⅡB型的致病基因位於染色體3q位點,發病年齡在青年期,表現為對稱性雙下肢的無力。HMSNⅡC型的致病基因不明確,發病年齡在兒童或青少年期,出現四肢遠端的肌無力伴膈肌和吞咽肌麻痹,一般沒有感覺障礙。HMSNⅡD型的致病基因位於染色體7p14位點,臨床表現為以上肢為主的遠端型肌肉無力。此家族的臨床、電生理和病理改變特點和HMSNⅡA型有相似之處,但老年前期發病和四肢同時出現無力在HMSNⅡA型比較少見。
檢查周圍神經的誘發電位波幅和感覺傳導速度對顯性遺傳性HMSNⅡ型的診斷具有重要意義。感覺和運動神經誘發電位波幅顯著下降是所有HMSNⅡ型病人的共同特點,而且波幅下降的幅度和大有髓神經纖維的脫失密切相關。和其他報導一樣,我們的資料也顯示神經傳導速度在這一個家族僅有輕度變慢,在HMSNⅡ型雖然感覺傳導速度減慢比運動傳導速度減慢更明顯,但傳導速度的下降一般不超過正常的60%。我們的結果證實電生理改變在同一個家族中的不同成員具有高度的一致性,電生理檢查可以發現家族中的亞臨床病人。
大直徑有髓神經纖維減少、出現再生神經纖維簇和缺乏典型洋蔥球樣結構是這個家族及其他文獻報導的顯性遺傳性HMSNⅡ型的主要病理特點,撕單根神經檢查一般沒有明顯的脫髓鞘和髓鞘再生改變,個別有髓神經纖維出現髓鞘變薄提示繼發性的脫髓鞘和髓鞘再形成。由於大有髓神經纖維的減少導致有髓神經纖維的直徑呈單峰分布,有髓神經纖維的再生導致小有髓神經纖維數目明顯增多,使有髓神經纖維的平均密度沒有明顯的減少。活動性的軸索病變很少出現,僅個別病人出現軸索腫大,巨大軸索可以出現在巨軸索神經病及其他許多周圍神經病,在更多的情況下為非特異性改變。
HMSNⅡ型的發病機理尚不明確。早期的研究結果提示最嚴重的病變部位在周圍神經的遠端,病變可能從遠端緩慢向近端發展。但Beciano的屍檢研究發現神經纖維的脫失、再生和軸索萎縮的嚴重程度在近端比遠端顯著,脊髓腰骶段的前角細胞和後根神經節的神經細胞出現脫失或萎縮。本研究病例發現肌束顫也提示病變累及前角細胞或其鄰近的軸索。最近Ono發現胸腰段脊髓的前角細胞出現萎縮和脫失,殘存的前角細胞樹突明顯減少,這些觀察提示病變部位可能在神經細胞核周體。
檢查
 神經炎
神經炎實驗室檢查:
血常規生化、腦脊液檢查均無異常發現。
其它輔助檢查:
1,肌電圖檢查可見運動神經及感覺神經傳導速度明顯減慢。
2,周圍神經活檢呈肥厚性間質性神經炎。
治療
1,本病無特殊治療方法,可套用維生素E維生素B1維生素B12、三磷腺苷(ATP)或CTP等治療維生素E胞磷膽鹼(胞二磷膽鹼)等也可試用.
2,加強鍛鍊針灸及理療對延緩病程可能有益。
預防
 神經炎
神經炎預後:
本病病程進展緩慢,時可加重或緩解,大多數患者可存活數十年。
對症治療可提高患者生活質量。
預防:
遺傳病治療困難,療效不滿意,預防顯得更為重要預防措施包括避免近親結婚,推行遺傳諮詢、攜帶者基因檢測及產前診斷和選擇性人工流產等防止患兒出生。
研究進展
(一)大多數CMT-1患者呈常染色體顯性遺傳傾向。孤立發病的CMT1一般為常染色體顯性遺傳的新突變。
 進行性肥厚性間質性神經炎
進行性肥厚性間質性神經炎CMT-1是至少有三個位點的遺傳異質性疾病:常見的CMT-1A位點位於17號染色體17p11.2區域,少見的CMT-1B位於1號染色體1q22-q23區域,第三類CMT-1C位點尚未定,但和17p11.2或1q22-q23均無關。71%CMT-1的患者與染色體17p11.2區域內的串聯複製(tandem-duplication)有關,除了極少例外複製均為同等大小即1.5Mb,提示在這一區域內有些序列影響這種持續的DNA重排。物理圖譜研究顯示1.5MbCMT-1A區域與串聯重複序列(tandem-repeat-sequence,-REPs)側接。CMT-1A複製很可能是近端和遠端CMT-1A-REPs的染色質錯排的結果。CMT-1A-REPs序列的三個拷貝位於CMT-1A複製染色體上,僅一個拷貝位於HNPP缺失染色體。1.5MbCMT-1A串聯複製含有PMP22基因,PMP22編碼22kD的蛋白,該蛋白是位於髓鞘緻密部的跨膜蛋白,功能尚不清。因為在CMT-1A複製患者可觀察到正常PMP22有三個完整拷貝,而大多數HNPP患者在同一區域僅有一個拷貝,現推測基因劑量是發病機制。CMT-1A中增加的PMP22基因拷貝數反映mRNA及蛋白表達的水平。在五個無血緣關係的非複製性CMT-1A家系患者發現了PMP22的五個不同顯性突變,這些PMP22突變導致輕與稍重度表現型。近期研究提示Thr(118)met突變可以是PMP22多態性,並不是所有PMP22突變都導致CMT1表現型。
CMT-1B型由1號染色體1q22-q23區域內的MPZ基因突變造成。MPZ編碼周圍髓鞘大部分豐富蛋白質,據結構推測可做為髓鞘兩個板層之間的粘附分子,有助於形成和維護髓鞘的緻密部分。在某些CMT-1B患者周圍神經活檢觀察鬆弛的髓鞘的結果支持這一假說。在36個無血緣關係的CMT1患者中有31個明顯MPZ突變,這些突變包括錯義突變、無義突變、剪接位點和移碼突變以及小缺失。有MPZ突變的CMT1患者如果表現為相對嚴重的CMT1表型一般比有1.5Mb串聯複製的CMT-1A表現更嚴重。
(二)X連鎖顯性遺傳性CMT1(CMTX)
起初認為CMTX很少見或不存在,後來明顯認識到CMTX可能被忽視了,因為傳遞的母親通常無症狀,而且X連鎖顯性遺傳CMT1的家庭經常歸在CMT-2,因為女性患者的電生理結果幾乎正常。
CMTX的位點位於X染色體q13-1區域,主要是Cx32基因的突變,Cx32編碼一種鑲嵌於各類細胞膜的缺口接頭(gap-junction)蛋白。Cx32蛋白的六個單位形成一個半通道或連線子。鄰近細胞細胞膜上的連線子形成功能性通道允許離子和小分子營養素通過。在周圍神經Cx32蛋白半通道位於節旁區和雪蘭氏間隙。CMT1患者Cx32的突變分析表明Cx32突變比以往想像的要頻繁,Cx32突變是CMT1患者第二大常見突變,僅次於CMT-1A的1.5Mb串聯複製。
(三)常染色體顯性遺傳型CMT2
CMT2可以是常染色體顯性遺傳或常染色體隱性遺傳。在一些典型HMSN-II表現型家庭已定位了常染色體顯性遺傳型CMT2的第一個位點(CMT2A)位於染色體1p35-p36。第二個位點(CMT2B)位於染色體3q13-q22。CMT2B家庭的患者有嚴重的遠端肢體無力,明顯感覺障礙及潰瘍,最終導致下肢遠端截肢。第三個位點(CMT2D)最近在一個大家系定位於染色體7p14。CMT2C位點尚未確定。
(四)DSS
DSS即HMSN-III或CMT-3目前已不再是有用的分型。該病1893年首先由DEJERINE和SOTTAS報導,對該型的描述差異較大,缺乏公認的診斷標準。最初DSS是指嬰兒或兒童發病、顯著無力為表現、嚴重的常染色體隱性遺傳的脫髓鞘性周圍神經病,病理所見為肥大神經及蔥球樣結構形成。後來還加了其他診斷指標如神經傳導速度低於6米/秒及腦脊液蛋白濃度升高者。基因分析顯示患者既可有PMP-22基因點突變(造成CMT-1A),又有P0髓鞘基因的點突變(造成CMT-1B)或有CMT-4的不同表現,或有EGR2基因突變。既往診斷為DSS的患者現可分為CMT-1A,CMT-1B,或CMT-4[12,13,14]。有時DSS也用做嚴重早髮型CMT1的同義詞。
(五)常染色體隱性遺傳型CMT-4
在近親結婚家庭已發現六種形式的常染色體隱性遺傳型CMT1的遺傳位點。第一個位點(CMT-4A)位於染色體8q13-q21,臨床表現較重,有些患者要靠輪椅行動。具有局部摺疊髓鞘的常染色體隱性遺傳型CMT1(CMT4B)的位點位於染色體11q23區。第三個位點(CMT4C)位於染色體5q23-q33。保加利亞吉普塞裔常染色體隱性遺傳型CMT1伴有耳聾(HMSNL),基因定位於染色體8q24(CMT4D)。CMT4E位點在EGR2基因內,CMT4F定位於19q13。
